Abstract
Cardiovascular disease, particularly ischemic heart disease, remains a leading cause of death worldwide. Although advances in pharmacological and device-based therapies have improved clinical outcomes, effective strategies for myocardial repair and regeneration remain limited. T-cadherin, a glycosylphosphatidylinositol-anchored atypical cadherin, has recently been identified as a functional receptor for both low-density lipoprotein cholesterol and adiponectin, a cardioprotective adipokine. Notably, the interaction between T-cadherin and adiponectin has emerged as a key regulator of exosome biogenesis and paracrine signaling within cardiovascular tissues. Exosomes are nanosized extracellular vesicles that carry protective molecular cargo, including microRNAs and proteins, and contribute to anti-inflammatory, antifibrotic, and angiogenic effects in the ischemic myocardium. However, their clinical translation is challenged by factors such as variability in yield, heterogeneity of exosome populations, and inefficient tissue targeting. Enhancing endogenous exosome production through the T-cadherin–adiponectin pathway may therefore offer a novel cell-free therapeutic strategy. This review explores the biological roles of T-cadherin and adiponectin in cardiovascular diseases, their regulatory influence on exosome formation, and the future potential of leveraging this axis for myocardial repair and regeneration.
-
Keywords: Adiponectin; Cardiovascular diseases; Extracellular vesicles; Regenerative medicine; T-cadherin
Introduction
Cardiovascular disease, particularly ischemic heart disease, remains the leading cause of morbidity and mortality worldwide, accounting for approximately 17.9 million deaths annually and 32% of all deaths [
1]. Despite major advances in pharmacological and interventional therapies, myocardial infarction continues to result in irreversible cardiomyocyte loss and fibrotic remodeling, reflecting the heart’s limited capacity for endogenous regeneration. This persistent limitation underscores the pressing need for novel strategies to restore myocardial structure and function.
Among emerging regenerative mechanisms, exosomes, which are small extracellular vesicles that mediate intercellular communication, have attracted increasing attention for their ability to deliver bioactive molecules, including microRNAs, lipids, and proteins. These vesicles are now widely recognized as key effectors of paracrine signaling that contribute to tissue repair and cardiovascular homeostasis.
T-cadherin, an atypical member of the cadherin superfamily, has emerged as an upstream regulator of exosome production and is uniquely anchored to the plasma membrane via a glycosylphosphatidylinositol (GPI) linkage. Unlike classical cadherins, T-cadherin lacks a transmembrane domain and functions primarily as a receptor rather than as a cell adhesion molecule.
T-cadherin binds selectively to high-molecular-weight (HMW) adiponectin, a cardioprotective adipokine, as well as to low-density lipoprotein (LDL), mediating distinct biological responses depending on the ligand involved. Upon adiponectin binding, T-cadherin facilitates its internalization into multivesicular bodies (MVBs), a process that stimulates the biogenesis and release of exosomes enriched with regenerative cargo [
2,
3]. These adiponectin-induced exosomes have been reported to exert anti-inflammatory, antifibrotic, and angiogenic effects in experimental models of ischemic injury [
4,
5].
Notably, T-cadherin deficiency abrogates the protective effects of adiponectin in cardiac tissues, underscoring the functional interdependence of this signaling axis [
5]. Furthermore, recent evidence suggests that both T-cadherin expression and circulating adiponectin levels are influenced by sex, genotype, and hormonal status, factors that may critically modulate the efficacy of exosome-based therapeutic approaches [
6,
7].
This review examines the structure and function of T-cadherin and adiponectin, their interactions within cardiovascular contexts, and the emerging concept of leveraging this axis to enhance endogenous exosome-mediated repair. By integrating recent molecular and translational evidence, the T-cadherin–adiponectin–exosome triad is proposed as a promising foundation for next-generation cell-free strategies aimed at myocardial regeneration.
Structural and functional integration of T-cadherin and adiponectin
Structure and ligand-binding properties of T-cadherin
T-cadherin (CDH13) is a structurally distinct member of the cadherin superfamily that lacks the canonical transmembrane and cytoplasmic domains present in classical cadherins. Instead, it is anchored to the outer leaflet of the plasma membrane through a GPI moiety, which allows it to function primarily in ligand binding and signal organization rather than in cell–cell adhesion [
8]. The extracellular domain of T-cadherin consists of 5 tandemly arranged cadherin repeats (EC1–EC5) that mediate homophilic dimerization as well as selective ligand binding. Structural modeling studies indicate that EC1 and EC2 are pivotal for ligand engagement and receptor conformational stability [
9,
10]. This unique architecture positions T-cadherin as a modulator of extracellular signaling, particularly in vascular and metabolic tissues.
Notably, T-cadherin selectively binds the HMW forms of adiponectin and exhibits markedly lower binding affinity for globular or low-molecular-weight isoforms, suggesting a preferential and functionally significant role in the retention and intracellular trafficking of the HMW isoform [
4].
In addition to adiponectin, T-cadherin also binds LDL. However, in contrast to the protective effects mediated by adiponectin, LDL–T-cadherin binding appears to promote pro-atherogenic oxidative stress and vascular dysfunction, particularly under atherosclerotic conditions [
11]. Furthermore, the interaction between LDL and T-cadherin is functionally distinct. Specific binding of LDL to T-cadherin can be competitively reduced by anti-apolipoprotein B antibodies, indicating that apolipoprotein B serves as a recognition site [
10]. Notably, LDL modifications, such as acetylation or carbamylation, do not significantly affect this interaction, in contrast to classical apolipoprotein B/E receptors. Moreover, the T-cadherin–LDL interaction is biologically active, promoting cell migration along LDL gradients and initiating mitogenic signaling in endothelial and epithelial cells through calcium mobilization, ERK1/2 phosphorylation, and nuclear translocation of NF-κB [
10]. This ligand-dependent functional dichotomy underscores the context-specific role of T-cadherin as a receptor for distinct ligands and as a signaling scaffold that modulates cellular outcomes in response to extracellular cues.
In addition to its dual role as a receptor for both HMW adiponectin and LDL, T-cadherin contributes to intracellular regulatory events, particularly those related to vesicle biology. Despite lacking a cytoplasmic tail, T-cadherin indirectly influences intracellular signaling by partitioning into lipid rafts, where it facilitates receptor clustering and signalosome assembly in coordination with proteins such as caveolin-1, a lipid raft–associated scaffolding protein [
12]. Through these interactions, T-cadherin may modulate key cellular processes, including endocytosis, ceramide efflux, and exosome biogenesis. Indeed, T-cadherin has been shown to spatially regulate MVB organization, a critical step in exosome release, particularly in response to adiponectin stimulation [
2,
3].
Clinically, T-cadherin is expressed in cardiovascular tissues, including endothelial cells, smooth muscle cells, and cardiomyocytes, and its expression is reduced in pathological conditions such as heart failure and atherosclerosis [
13,
14].
Adiponectin is an adipocyte-derived hormone that circulates at high concentrations and exerts broad protective effects on the cardiovascular system. It exists in several multimeric forms, including low-molecular-weight trimers, middle-molecular-weight hexamers, and HMW multimers. Among these isoforms, the HMW form is considered the most biologically active in cardiovascular tissues, particularly in anti-inflammatory, insulin-sensitizing, and anti-atherogenic contexts [
15-
17].
During puberty, a notable decrease in circulating adiponectin levels is observed in males. In addition, adiponectin levels are known to increase with advancing age. A circadian variation of approximately 20% in adiponectin concentration occurs over a 24-hour period, with levels declining overnight and reaching their nadir in the early morning. This diurnal fluctuation is more pronounced in females than in males, but it does not appear to differ according to obesity status [
18].
Adiponectin exerts its effects through 3 main receptors: AdipoR1, AdipoR2, and T-cadherin [
14]. AdipoR1 is predominantly expressed in skeletal muscle and exhibits high affinity for globular adiponectin, whereas AdipoR2 is primarily expressed in the liver and mediates peroxisome proliferator-activated receptor-α (PPARα)–dependent signaling pathways [
19]. In contrast, T-cadherin binds specifically to the HMW form of adiponectin and is highly expressed in cardiovascular tissues, including endothelial cells lining blood vessels, cardiomyocytes, smooth muscle cells, and the microvascular endothelium of skeletal muscle and the aorta [
5,
20]. This tissue distribution supports a central role for T-cadherin in vascular homeostasis and cardiac remodeling [
20]. Unlike circulating adiponectin levels, adiponectin receptor expression in adipose tissue does not differ between sexes. However, in contrast to adipose tissue, adiponectin receptors are more highly expressed in the skeletal muscle of males. Interestingly, circulating adiponectin levels are lower in males than in females, a pattern that may be partially explained by increased receptor expression in male skeletal muscle [
18].
The 2 ligands for T-cadherin, adiponectin and LDL, are both large molecular complexes of comparable size. LDL particles are roughly spherical, with diameters of approximately 18–25 nm, whereas HMW adiponectin forms fan-shaped or compact, bunch-like structures, with globular domains extending up to 25–32 nm [
14]. This structural similarity suggests the possibility of direct competition between LDL and HMW adiponectin for binding to T-cadherin [
10].
This competition has important clinical implications because ligand concentrations fluctuate under physiological and pathological conditions. Under normal physiological conditions, the circulating concentration of LDL (approximately 0.6 mg/mL, protein basis) and adiponectin (approximately 1–30 μg/mL, representing levels 103–106-fold higher than those of typical hormones and cytokines) are relatively balanced [
21]. However, in pathological states such as metabolic syndrome, LDL levels can rise above 2 mg/mL, while adiponectin concentrations may fall below 1 μg/mL.
This imbalance shifts the ligand ratio by more than tenfold in favor of LDL, potentially leading to the competitive displacement of cardioprotective adiponectin by atherogenic LDL at the level of T-cadherin binding [
10]. Affinity data support this dynamic, as the dissociation constant of LDL for T-cadherin is approximately 40 μg/mL, whereas HMW adiponectin achieves half-maximal binding at a substantially lower concentration of approximately 2.2 μg/mL.
Hypoadiponectinemia, commonly defined as plasma adiponectin levels below 4 μg/mL, has been associated with impaired glucose and lipid metabolism and an increased risk of coronary artery disease, particularly in men without a prior history of cardiovascular disease [
14,
17]. Adiponectin exhibits clear sexual dimorphism, with women consistently displaying higher circulating levels than men [
22]. Notably, menopause does not appear to affect adiponectin levels, and neither oophorectomy nor estrogen replacement therapy alters plasma adiponectin concentrations in women [
22]. Similarly, oophorectomy in female mice does not result in significant changes in adiponectin levels [
23]. In contrast, castration of male mice leads to a marked increase in adiponectin levels [
24]. Likewise, in cell culture models, testosterone treatment reduces both circulating adiponectin levels and adiponectin secretion while simultaneously inducing insulin resistance. Collectively, these findings suggest that adiponectin deficiency may contribute to the elevated cardiovascular risk observed in men and that sex-based biological differences modulate the T-cadherin–adiponectin signaling axis [
20].
In addition to sex-related hormonal and receptor differences, previous studies have demonstrated that plasma adiponectin levels are inversely correlated with body mass index and positively associated with age, indicating that metabolic and demographic variables jointly shape adiponectin signaling [
25,
26]. Therefore, a comprehensive framework incorporating sex, body mass index, and age is essential for elucidating the pathophysiological role of the T-cadherin–adiponectin axis and for developing more precise strategies for cardiovascular disease prevention and treatment.
Upon binding to T-cadherin on the surface of cardiovascular cells, particularly endothelial cells and cardiomyocytes, HMW adiponectin initiates a noncanonical yet highly effective cardioprotective pathway. Unlike AdipoR1 and AdipoR2, which signal through intracellular phosphorylation cascades such as AMPK and PPARα, the T-cadherin–adiponectin axis does not involve direct enzymatic signaling within the cytoplasm. Instead, it depends on the biophysical clustering of GPI-anchored T-cadherin within lipid rafts, leading to recruitment of endocytic machinery and subsequent formation of MVBs [
2].
These MVBs subsequently mature and release exosomes and nanovesicles containing diverse bioactive cargo, including angiogenic proteins, lipids, and microRNAs such as miR-126 and miR-21. Once secreted, these exosomes are taken up by neighboring or distant cardiovascular cells, thereby mediating paracrine effects that promote endothelial repair, stimulate capillary growth, attenuate proinflammatory signaling, and suppress myocardial fibrosis [
27,
28]. This exosome-driven paracrine communication plays a critical role in maintaining vascular homeostasis, limiting infarct size during ischemic events, and facilitating cardioprotection [
3,
5,
28].
The importance of this pathway is further underscored by experimental findings showing that deletion of the T-cadherin gene abolishes the tissue-binding and cardioprotective functions of adiponectin, despite adequate systemic levels of the hormone [
2,
4]. Thus, T-cadherin functions as a molecular gatekeeper for adiponectin-induced exosomal signaling in the heart.
In addition to promoting exosome biogenesis through its interaction with T-cadherin, adiponectin exerts a distinct cardioprotective effect by directly inhibiting atherogenic LDL. Adiponectin interacts with modified LDL species, including oxidized LDL (oxLDL) and L5, forming molecular complexes that effectively neutralize their pathogenic activity. This neutralization occurs at physiological adiponectin concentrations and is independent of classical adiponectin receptor–mediated signaling pathways. Notably, adiponectin-bound oxLDL fails to trigger downstream inflammatory responses, including NF-κB activation and oxidative stress. Together, these findings identify an additional anti-atherogenic mechanism of adiponectin that involves direct ligand neutralization, as demonstrated in both in vitro and in vivo models [
29].
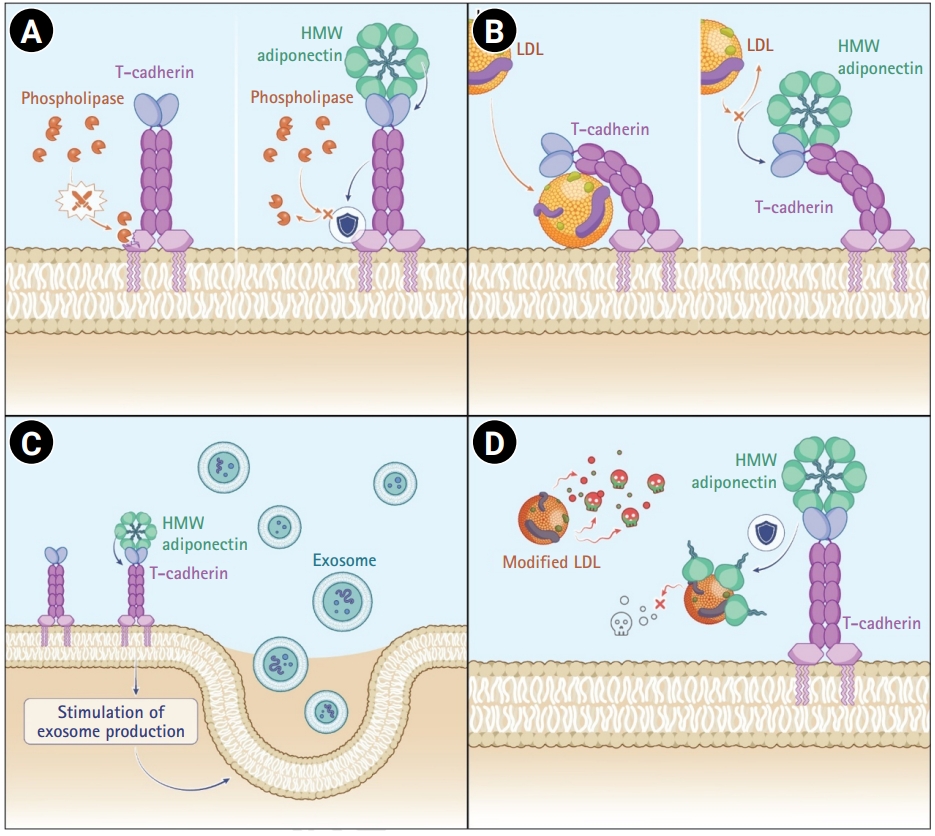
Adiponectin also stabilizes T-cadherin protein on the plasma membrane by inhibiting its cleavage by GPI-specific phospholipases and is likely to prevent T-cadherin propeptide cleavage [
10,
21] (
Fig. 1A). In T-cadherin-deficient mice, adiponectin fails to associate with tissues and instead accumulates in the circulation, thereby mimicking the cardiovascular phenotype observed in adiponectin knockout models. Enzymatic cleavage of T-cadherin using phosphatidylinositol-specific phospholipase C increases plasma adiponectin levels while reducing tissue-bound adiponectin. In vivo animal studies further demonstrate that both circulating and tissue-bound adiponectin levels depend on T-cadherin expression. Thus, T-cadherin acts as a critical modulator of adiponectin distribution between the bloodstream and peripheral tissues. Moreover, adiponectin positively regulates T-cadherin protein expression, likely by suppressing enzymatic cleavage of T-cadherin, thereby establishing a positive feedback loop that reinforces adiponectin binding to T-cadherin at the plasma membrane [
21].
Thus, adiponectin confers cardiovascular protection through 4 complementary mechanisms (
Fig. 1): (1) stabilization of membrane-anchored T-cadherin through inhibition of GPI-anchor cleavage by phospholipases and regulation of propeptide processing to promote surface retention of the pro-form; (2) competitive binding against atherogenic LDL for T-cadherin engagement; (3) stimulation of cardioprotective exosome production via interaction with T-cadherin; and (4) direct neutralization of modified LDL species through molecular complex formation. Collectively, these pleiotropic actions position adiponectin as a central regulator of vascular homeostasis and a promising therapeutic target in atherosclerotic cardiovascular disease.
Cardiovascular clinical implications of the T-cadherin–adiponectin axis
Atherosclerosis
T-cadherin modulates endothelial responses to LDL, while adiponectin directly binds oxLDL [
29,
30]. Together, T-cadherin and adiponectin inhibit foam cell formation, reduce vascular inflammation, and modulate lipid uptake within the arterial wall [
31,
32]. Elevated plasma LDL levels combined with reduced adiponectin concentrations are characteristic features of atherosclerosis, dyslipidemia, and metabolic syndrome [
14]. Genetic models deficient in T-cadherin exhibit enhanced atherogenesis, even in the presence of normal circulating adiponectin levels [
5].
Genetic investigations and experimental studies in preclinical models have provided robust evidence supporting the cardioprotective role of the T-cadherin–adiponectin axis in atherosclerosis. Mouse models lacking T-cadherin develop larger and more advanced atherosclerotic plaques with increased inflammatory cell infiltration, even when circulating adiponectin levels are preserved [
5]. Similarly, adiponectin-knockout mice exhibit exaggerated lipid accumulation and heightened vascular inflammation [
4].
Genome-wide association studies (GWAS) further support these observations. Polymorphisms in
CDH13 (e.g., rs3865188) and
ADIPOQ (e.g., rs266729) are associated with interindividual variation in adiponectin levels and susceptibility to coronary artery disease [
33,
34]. A pilot study demonstrated that carotid intima-media thickness is inversely correlated with HMW adiponectin levels in males, and that the G allele of the rs12444338 single-nucleotide polymorphism in the
CDH13 gene is associated with lower circulating T-cadherin levels and reduced intima-media thickness, a profile that may be considered atheroprotective [
35].
Collectively, these findings highlight the dual role of T-cadherin as a receptor for both adiponectin and atherogenic LDL and underscore the importance of adiponectin bioavailability, mediated by its binding to T-cadherin and its capacity to suppress oxidized LDL, in regulating atherogenic processes. Together, these insights support the T-cadherin–adiponectin axis as a promising target for preventive and therapeutic strategies in cardiovascular disease.
Myocardial injury and fibrosis
Animal models of myocardial infarction have demonstrated that T-cadherin is essential for local adiponectin-mediated cardioprotection. In the absence of T-cadherin, infarct size increases, neovascularization is suppressed, and ventricular function deteriorates as a result of impaired AMPK activation and increased myocardial apoptosis [
5]. T-cadherin and adiponectin also regulate fibroblast activation and extracellular matrix remodeling. When this axis is downregulated, myocardial fibrosis and left ventricular hypertrophy are exacerbated, thereby contributing to the development and progression of heart failure [
13]. Furthermore, T-cadherin is required for adiponectin-induced vascular regeneration, as demonstrated in hindlimb ischemia models, in which T-cadherin deficiency abolishes revascularization despite preserved adiponectin levels [
36]. These findings indicate that T-cadherin mediates the acute cardioprotective effects of adiponectin and plays a central role in long-term structural remodeling following myocardial injury.
Therapeutic modulation of adiponectin and T-cadherin pathway
Lifestyle interventions: exercise and metabolic modulation
Regular endurance exercise and caloric modulation have been implicated in regulating the adiponectin–T-cadherin axis. Circulating adiponectin levels are responsive to lifestyle interventions. A comprehensive meta-analysis demonstrated that aerobic exercise significantly increases circulating adiponectin levels in prediabetic and diabetic adults, independent of weight loss [
37]. T-cadherin expression is likewise sensitive to metabolic cues and lifestyle factors. In murine models, caloric overload followed by caloric restriction has been shown to upregulate both adiponectin and T-cadherin expression in cardiac tissues. This nutritional modulation is accompanied by improvements in mitochondrial function, enhanced AMPK and endothelial nitric oxide synthase activity, and attenuation of oxidative stress markers, highlighting a potentially synergistic mechanism through which energy balance influences the adiponectin–T-cadherin axis [
38].
Among pharmacological agents, thiazolidinediones such as pioglitazone are known to significantly increase adiponectin levels, particularly the HMW isoform, thereby improving insulin sensitivity and endothelial function [
31]. In addition, sodium–glucose cotransporter 2 inhibitors, including empagliflozin, have been associated with elevated adiponectin concentrations, which may partially explain the cardiovascular benefits observed in patients with heart failure and diabetes [
39]. In contrast, statins exert variable effects on adiponectin levels. Pravastatin does not alter adiponectin levels in healthy individuals but has been shown to increase plasma adiponectin concentrations in patients with impaired glucose tolerance or coronary artery disease [
40]. Conversely, rosuvastatin significantly reduces both total and HMW adiponectin levels [
41], whereas fluvastatin, atorvastatin, and simvastatin exhibit minimal or inconsistent effects [
19].
Given the anti-inflammatory and insulin-sensitizing properties of adiponectin, such variability may partially explain the differential effects of statins on metabolic syndrome and cardiovascular outcomes. Alterations in adiponectin levels may influence atheroprotection and metabolic control, with important implications for precision medicine. Taken together, pharmacological strategies that increase adiponectin levels may enhance T-cadherin-mediated cardioprotective pathways, particularly in patients with hypoadiponectinemia or metabolic syndrome.
Although no approved therapy directly targets CDH13, several pharmacological agents may indirectly modulate its expression. Recent studies have shown that pharmacological activators of hypoxia-inducible factor-1 (HIF-1), including roxadustat and daprodustat, significantly upregulate T-cadherin expression in vitro. These agents enhance CDH13 transcription in murine endothelial cells under hypoxic conditions, suggesting that HIF-1 activation may represent a viable therapeutic strategy for increasing T-cadherin expression in ischemic cardiovascular settings [
42].
In addition to endogenous modulation, preclinical studies have highlighted the therapeutic potential of directly augmenting T-cadherin expression through gene delivery approaches. Notably, T-cadherin appears to be essential for adiponectin-induced exosome biogenesis, providing a mechanistic link between receptor function and downstream regenerative signaling cascades [
2]. These exosomes carry microRNAs and proteins that mediate angiogenesis, exert antifibrotic effects, and promote cardiomyocyte survival, thereby representing a promising therapeutic avenue for myocardial repair [
2,
3]. Upregulation of T-cadherin in cardiovascular tissues has been associated with increased extracellular vesicle release, suggesting a potential role for this pathway in myocardial repair following ischemic injury [
2,
5,
42].
However, the direct causal relationship between cardiac-specific overexpression of T-cadherin and enhanced myocardial recovery—particularly through extracellular vesicle–mediated mechanisms—has not yet been fully elucidated. Further in vivo studies employing targeted gene delivery systems, such as adeno-associated viral vectors, are warranted to validate this mechanism and to explore its therapeutic potential in ischemic heart disease. Collectively, these findings support a multifaceted strategy for enhancing T-cadherin activity, encompassing both systemic metabolic modulation and targeted gene-based interventions. Further research is required to define tissue-specific effects, long-term safety, and the translational feasibility of these approaches in humans.
Future perspectives
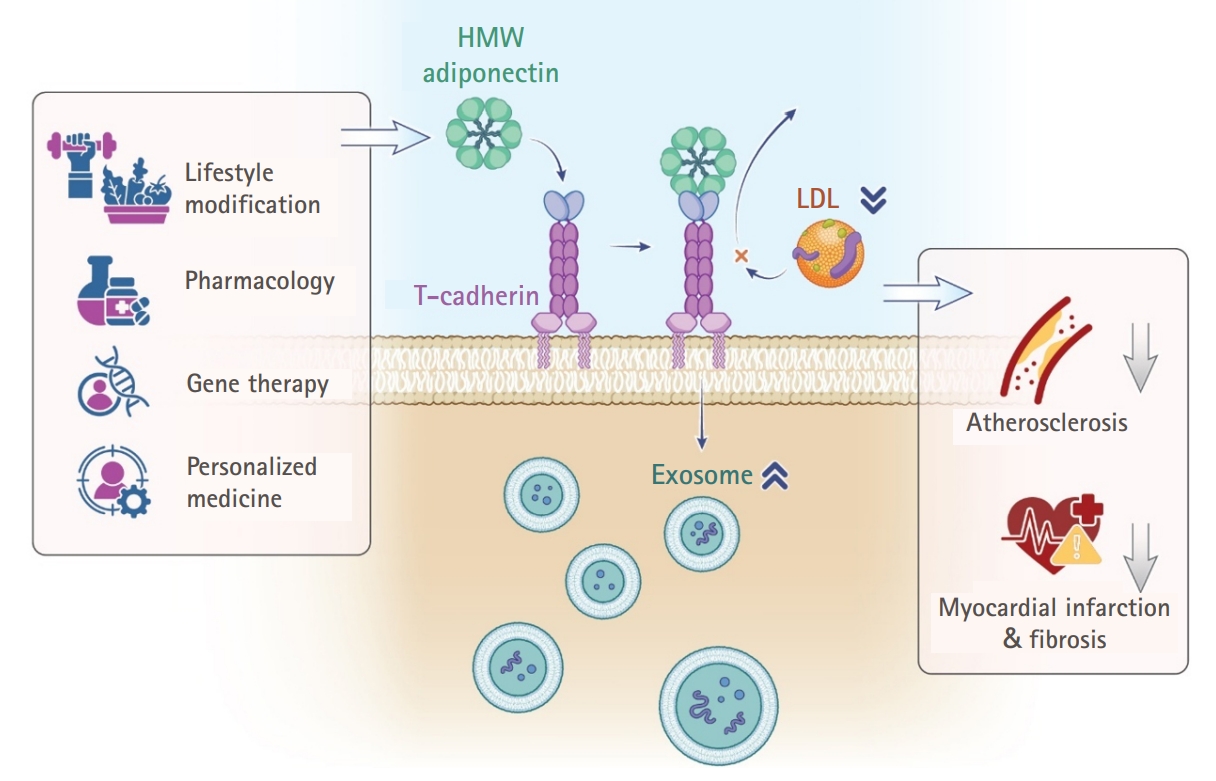
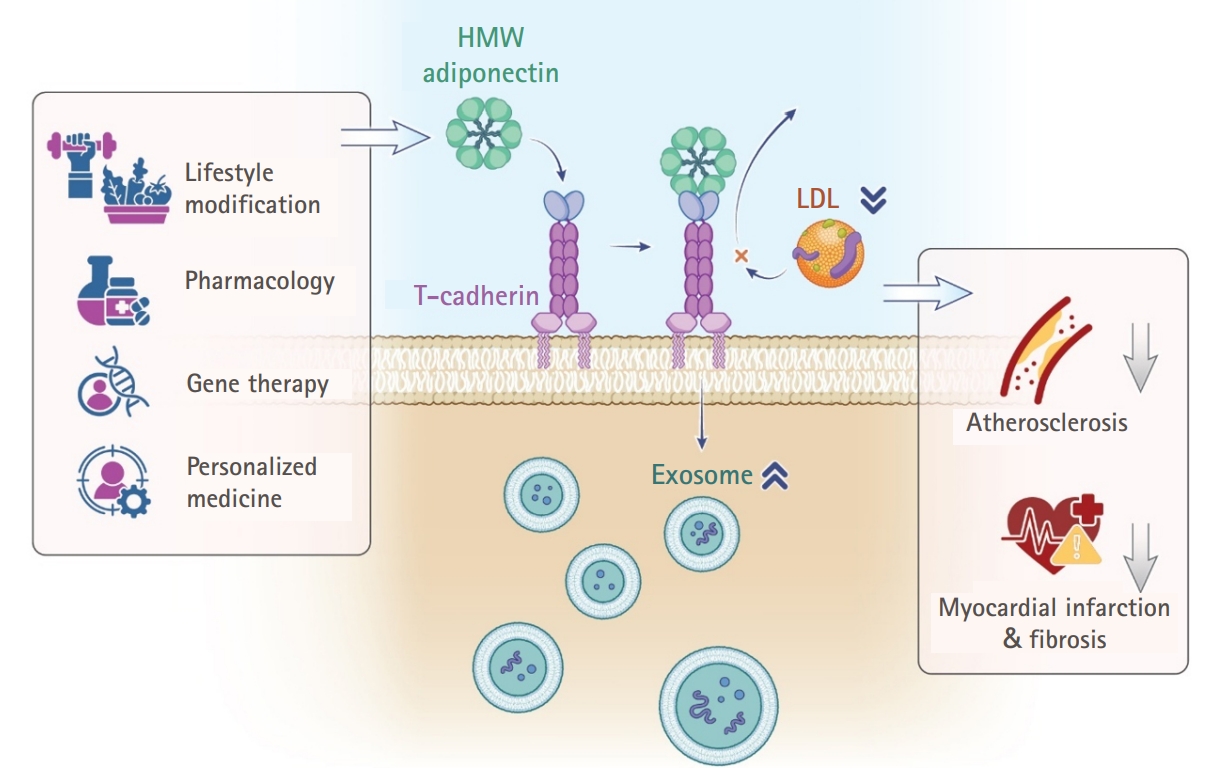
Targeted modulation of the adiponectin–T-cadherin–exosome axis holds substantial promise in regenerative cardiology. Future investigations should explore pharmacological, gene therapy, and biomimetic strategies to harness this pathway within personalized medicine frameworks (
Fig. 2). Given the influence of sex and hormonal status on adiponectin levels and T-cadherin signaling, therapeutic strategies may require stratification according to sex and age. Women, who typically exhibit higher baseline adiponectin levels in association with estrogen, may derive greater benefit from interventions that enhance T-cadherin signaling, whereas men, who generally have lower adiponectin levels, may require approaches that augment adiponectin availability or improve receptor sensitivity. Engineering biomimetic exosomes or T-cadherin–overexpressing stem cells represents another potential strategy to enhance myocardial repair. Furthermore, leveraging genetic polymorphism data related to T-cadherin may facilitate precision medicine approaches for cardiovascular regeneration.
Conclusion
In conclusion, the T-cadherin–adiponectin axis represents a novel and physiologically relevant mechanism of cardiovascular protection, particularly through its role in promoting exosome biogenesis and paracrine regenerative signaling. Although adiponectin is a well-established cardioprotective adipokine, the identification of T-cadherin as its nonclassical receptor and regulator of vesicular trafficking has opened new avenues for cell-free therapeutic strategies. Harnessing or mimicking this axis through pharmacological agents, genetic modulation, or biomimetic exosome delivery offers a promising approach to myocardial regeneration following infarction or in chronic heart failure.
-
Authors’ contribution
All the work was done by In Sook Kang.
-
Conflict of interest
No potential conflict of interest relevant to this article was reported.
-
Funding
This work was supported by the Ewha Womans University Research Grant of 2023 (1-2023-1729-001-1).
-
Data availability
Not applicable.
-
Acknowledgments
None.
-
Supplementary materials
None.
Fig. 1.High-molecular-weight (HMW) adiponectin–T-cadherin mechanism: 4 core cardioprotective effects. HMW adiponectin exerts 4 major actions: (A) stabilizing membrane-anchored T-cadherin by inhibiting phospholipase-mediated glycosylphosphatidylinositol (GPI)-anchor cleavage; (B) acting as a competitive ligand against atherogenic low-density lipoprotein (LDL) for T-cadherin binding; (C) promoting cardioprotective exosome production through engagement with T-cadherin; and (D) directly neutralizing the harmful effects of modified LDL through molecular complex formation.
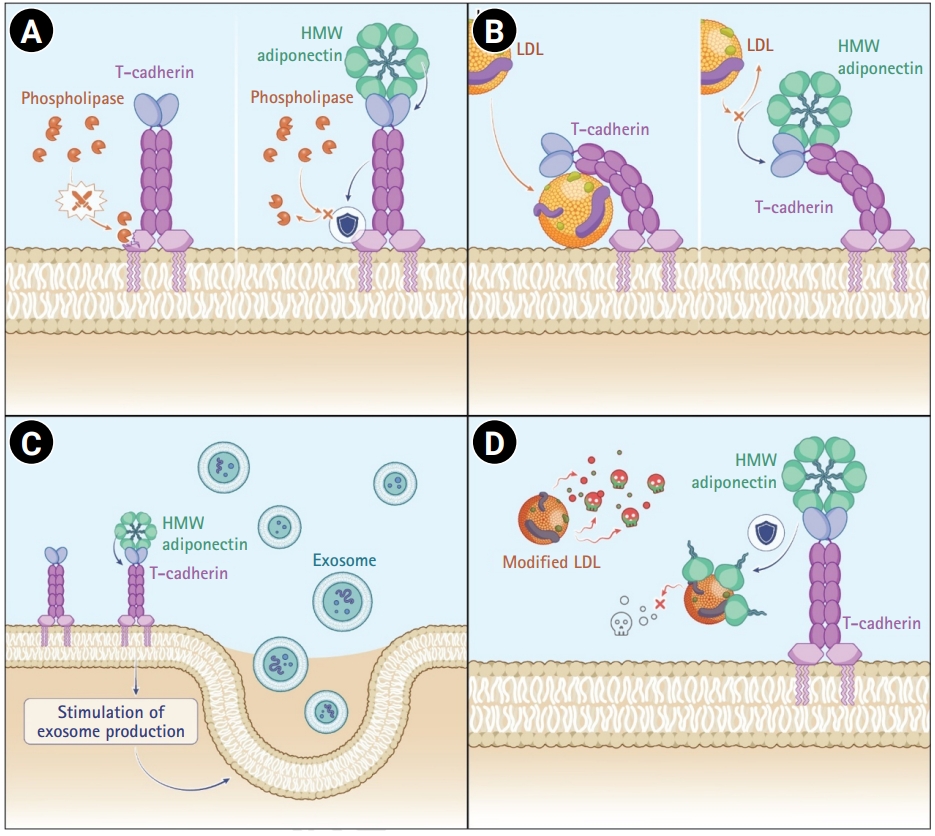
Fig. 2.Integrated overview of high-molecular-weight (HMW) adiponectin–T-cadherin–mediated cardiometabolic protection. This figure summarizes the overall conceptual framework of the study. Centrally, HMW adiponectin binding to T-cadherin promotes cardioprotective exosome biogenesis and attenuates low-density lipoprotein cholesterol (LDL-C)–driven atherogenic signaling. On the left, lifestyle modification, pharmacological interventions, gene-based strategies, and personalized medicine approaches are depicted as upstream modulators that increase circulating HMW adiponectin levels, strengthen T-cadherin interactions, and amplify exosome production. On the right, the downstream consequences of this enhanced signaling include suppression of LDL-C accumulation, attenuation of atherosclerotic plaque progression, reduction of myocardial infarction–related injury, and mitigation of post-ischemic fibrosis.
References
- 1. World Health Organization (WHO). Cardiovascular diseases (CVDs): key facts [Internet]. WHO; 2025 [cited 2025 Jul 31]. Available from: https://www.who.int/news-room/fact-sheets/detail/cardiovascular-diseases-(cvds)
- 2. Obata Y, Kita S, Koyama Y, Fukuda S, Takeda H, Takahashi M, Fujishima Y, Nagao H, Masuda S, Tanaka Y, Nakamura Y, Nishizawa H, Funahashi T, Ranscht B, Izumi Y, Bamba T, Fukusaki E, Hanayama R, Shimada S, Maeda N, Shimomura I. Adiponectin/T-cadherin system enhances exosome biogenesis and decreases cellular ceramides by exosomal release. JCI Insight 2018;3:e99680. https://doi.org/10.1172/jci.insight.99680
- 3. Kita S, Shimomura I. Stimulation of exosome biogenesis by adiponectin, a circulating factor secreted from adipocytes. J Biochem 2021;169:173-179. https://doi.org/10.1093/jb/mvaa105
- 4. Fukuoka K, Mineo R, Kita S, Fukuda S, Okita T, Kawada-Horitani E, Iioka M, Fujii K, Kawada K, Fujishima Y, Nishizawa H, Maeda N, Shimomura I. ER stress decreases exosome production through adiponectin/T-cadherin-dependent and -independent pathways. J Biol Chem 2023;299:105114. https://doi.org/10.1016/j.jbc.2023.105114
- 5. Denzel MS, Scimia MC, Zumstein PM, Walsh K, Ruiz-Lozano P, Ranscht B. T-cadherin is critical for adiponectin-mediated cardioprotection in mice. J Clin Invest 2010;120:4342-4352. https://doi.org/10.1172/JCI43464
- 6. Filippi E, Sentinelli F, Romeo S, Arca M, Berni A, Tiberti C, Verrienti A, Fanelli M, Fallarino M, Sorropago G, Baroni MG. The adiponectin gene SNP+276G>T associates with early-onset coronary artery disease and with lower levels of adiponectin in younger coronary artery disease patients (age <or=50 years). J Mol Med (Berl) 2005;83:711-719. https://doi.org/10.1007/s00109-005-0667-z
- 7. Kitamoto A, Kitamoto T, Nakamura T, Matsuo T, Nakata Y, Hyogo H, Ochi H, Kamohara S, Miyatake N, Kotani K, Mineo I, Wada J, Ogawa Y, Yoneda M, Nakajima A, Funahashi T, Miyazaki S, Tokunaga K, Masuzaki H, Ueno T, Chayama K, Hamaguchi K, Yamada K, Hanafusa T, Oikawa S, Sakata T, Tanaka K, Matsuzawa Y, Hotta K. CDH13 polymorphisms are associated with adiponectin levels and metabolic syndrome traits independently of visceral fat mass. J Atheroscler Thromb 2016;23:309-319. https://doi.org/10.5551/jat.31567
- 8. Ciatto C, Bahna F, Zampieri N, VanSteenhouse HC, Katsamba PS, Ahlsen G, Harrison OJ, Brasch J, Jin X, Posy S, Vendome J, Ranscht B, Jessell TM, Honig B, Shapiro L. T-cadherin structures reveal a novel adhesive binding mechanism. Nat Struct Mol Biol 2010;17:339-347. https://doi.org/10.1038/nsmb.1781
- 9. Fukuda S, Kita S, Obata Y, Fujishima Y, Nagao H, Masuda S, Tanaka Y, Nishizawa H, Funahashi T, Takagi J, Maeda N, Shimomura I. The unique prodomain of T-cadherin plays a key role in adiponectin binding with the essential extracellular cadherin repeats 1 and 2. J Biol Chem 2017;292:7840-7849. https://doi.org/10.1074/jbc.M117.780734
- 10. Balatskaya MN, Balatskii AV, Sharonov GV, Tkachuk VA. T-cadherin as a novel receptor regulating metabolism in the blood vessel and heart cells: from structure to function. J Evol Biochem Physiol 2016;52:103-118. https://doi.org/10.1134/S0022093016020010
- 11. Balatskaya MN, Sharonov GV, Baglay AI, Rubtsov YP, Tkachuk VA. Different spatiotemporal organization of GPI-anchored T-cadherin in response to low-density lipoprotein and adiponectin. Biochim Biophys Acta Gen Subj 2019;1863:129414. https://doi.org/10.1016/j.bbagen.2019.129414
- 12. Philippova MP, Bochkov VN, Stambolsky DV, Tkachuk VA, Resink TJ. T-cadherin and signal-transducing molecules co-localize in caveolin-rich membrane domains of vascular smooth muscle cells. FEBS Lett 1998;429:207-210. https://doi.org/10.1016/s0014-5793(98)00598-5
- 13. Baltrūnienė V, Rinkūnaitė I, Bogomolovas J, Bironaitė D, Kažukauskienė I, Šimoliūnas E, Ručinskas K, Puronaitė R, Bukelskienė V, Grabauskienė AV. The role of cardiac T-cadherin in the indicating heart failure severity of patients with non-ischemic dilated cardiomyopathy. Medicina (Kaunas) 2020;56:27. https://doi.org/10.3390/medicina56010027
- 14. Rubina KA, Semina EV, Kalinina NI, Sysoeva VY, Balatskiy AV, Tkachuk VA. Revisiting the multiple roles of T-cadherin in health and disease. Eur J Cell Biol 2021;100:151183. https://doi.org/10.1016/j.ejcb.2021.151183
- 15. Waki H, Yamauchi T, Kamon J, Ito Y, Uchida S, Kita S, Hara K, Hada Y, Vasseur F, Froguel P, Kimura S, Nagai R, Kadowaki T. Impaired multimerization of human adiponectin mutants associated with diabetes: molecular structure and multimer formation of adiponectin. J Biol Chem 2003;278:40352-40363. https://doi.org/10.1074/jbc.M300365200
- 16. Basu R, Pajvani UB, Rizza RA, Scherer PE. Selective downregulation of the high molecular weight form of adiponectin in hyperinsulinemia and in type 2 diabetes: differential regulation from nondiabetic subjects. Diabetes 2007;56:2174-2177. https://doi.org/10.2337/db07-0185
- 17. Maeda N, Funahashi T, Matsuzawa Y, Shimomura I. Adiponectin, a unique adipocyte-derived factor beyond hormones. Atherosclerosis 2020;292:1-9. https://doi.org/10.1016/j.atherosclerosis.2019.10.021
- 18. Nguyen TM. Adiponectin: role in physiology and pathophysiology. Int J Prev Med 2020;11:136. https://doi.org/10.4103/ijpvm.IJPVM_193_20
- 19. Peng J, Chen Q, Wu C. The role of adiponectin in cardiovascular disease. Cardiovasc Pathol 2023;64:107514. https://doi.org/10.1016/j.carpath.2022.107514
- 20. Clark JL, Taylor CG, Zahradka P. Exploring the cardio-metabolic relevance of T-cadherin: a pleiotropic adiponectin receptor. Endocr Metab Immune Disord Drug Targets 2017;17:200-206. https://doi.org/10.2174/1871530317666170818120224
- 21. Matsuda K, Fujishima Y, Maeda N, Mori T, Hirata A, Sekimoto R, Tsushima Y, Masuda S, Yamaoka M, Inoue K, Nishizawa H, Kita S, Ranscht B, Funahashi T, Shimomura I. Positive feedback regulation between adiponectin and T-cadherin impacts adiponectin levels in tissue and plasma of male mice. Endocrinology 2015;156:934-946. https://doi.org/10.1210/en.2014-1618
- 22. Combs TP, Berg AH, Rajala MW, Klebanov S, Iyengar P, Jimenez-Chillaron JC, Patti ME, Klein SL, Weinstein RS, Scherer PE. Sexual differentiation, pregnancy, calorie restriction, and aging affect the adipocyte-specific secretory protein adiponectin. Diabetes 2003;52:268-276. https://doi.org/10.2337/diabetes.52.2.268
- 23. Gui Y, Silha JV, Murphy LJ. Sexual dimorphism and regulation of resistin, adiponectin, and leptin expression in the mouse. Obes Res 2004;12:1481-1491. https://doi.org/10.1038/oby.2004.185
- 24. Nishizawa H, Shimomura I, Kishida K, Maeda N, Kuriyama H, Nagaretani H, Matsuda M, Kondo H, Furuyama N, Kihara S, Nakamura T, Tochino Y, Funahashi T, Matsuzawa Y. Androgens decrease plasma adiponectin, an insulin-sensitizing adipocyte-derived protein. Diabetes 2002;51:2734-2741. https://doi.org/10.2337/diabetes.51.9.2734
- 25. Kim-Mitsuyama S, Soejima H, Yasuda O, Node K, Jinnouchi H, Yamamoto E, Sekigami T, Ogawa H, Matsui K. Total adiponectin is associated with incident cardiovascular and renal events in treated hypertensive patients: subanalysis of the ATTEMPT-CVD randomized trial. Sci Rep 2019;9:16589. https://doi.org/10.1038/s41598-019-52977-x
- 26. Arita Y, Kihara S, Ouchi N, Takahashi M, Maeda K, Miyagawa J, Hotta K, Shimomura I, Nakamura T, Miyaoka K, Kuriyama H, Nishida M, Yamashita S, Okubo K, Matsubara K, Muraguchi M, Ohmoto Y, Funahashi T, Matsuzawa Y. Paradoxical decrease of an adipose-specific protein, adiponectin, in obesity. Biochem Biophys Res Commun 1999;257:79-83. https://doi.org/10.1006/bbrc.1999.0255
- 27. Kita S, Maeda N, Shimomura I. Interorgan communication by exosomes, adipose tissue, and adiponectin in metabolic syndrome. J Clin Invest 2019;129:4041-4049. https://doi.org/10.1172/JCI129193
- 28. Tanaka Y, Kita S, Nishizawa H, Fukuda S, Fujishima Y, Obata Y, Nagao H, Masuda S, Nakamura Y, Shimizu Y, Mineo R, Natsukawa T, Funahashi T, Ranscht B, Fukada SI, Maeda N, Shimomura I. Adiponectin promotes muscle regeneration through binding to T-cadherin. Sci Rep 2019;9:16. https://doi.org/10.1038/s41598-018-37115-3
- 29. Kakino A, Fujita Y, Ke LY, Chan HC, Tsai MH, Dai CY, Chen CH, Sawamura T. Adiponectin forms a complex with atherogenic LDL and inhibits its downstream effects. J Lipid Res 2021;62:100001. https://doi.org/10.1194/jlr.RA120000767
- 30. Philippova M, Suter Y, Toggweiler S, Schoenenberger AW, Joshi MB, Kyriakakis E, Erne P, Resink TJ. T-cadherin is present on endothelial microparticles and is elevated in plasma in early atherosclerosis. Eur Heart J 2011;32:760-771. https://doi.org/10.1093/eurheartj/ehq206
- 31. Yamauchi T, Kamon J, Waki H, Terauchi Y, Kubota N, Hara K, Mori Y, Ide T, Murakami K, Tsuboyama-Kasaoka N, Ezaki O, Akanuma Y, Gavrilova O, Vinson C, Reitman ML, Kagechika H, Shudo K, Yoda M, Nakano Y, Tobe K, Nagai R, Kimura S, Tomita M, Froguel P, Kadowaki T. The fat-derived hormone adiponectin reverses insulin resistance associated with both lipoatrophy and obesity. Nat Med 2001;7:941-946. https://doi.org/10.1038/90984
- 32. Ouchi N, Kihara S, Arita Y, Nishida M, Matsuyama A, Okamoto Y, Ishigami M, Kuriyama H, Kishida K, Nishizawa H, Hotta K, Muraguchi M, Ohmoto Y, Yamashita S, Funahashi T, Matsuzawa Y. Adipocyte-derived plasma protein, adiponectin, suppresses lipid accumulation and class A scavenger receptor expression in human monocyte-derived macrophages. Circulation 2001;103:1057-1063. https://doi.org/10.1161/01.cir.103.8.1057
- 33. Dastani Z, Hivert MF, Timpson N, Perry JR, Yuan X, Scott RA, Henneman P, Heid IM, Kizer JR, Lyytikäinen LP, Fuchsberger C, Tanaka T, Morris AP, Small K, Isaacs A, Beekman M, Coassin S, Lohman K, Qi L, Kanoni S, Pankow JS, Uh HW, Wu Y, Bidulescu A, Rasmussen-Torvik LJ, Greenwood CM, Ladouceur M, Grimsby J, Manning AK, Liu CT, Kooner J, Mooser VE, Vollenweider P, Kapur KA, Chambers J, Wareham NJ, Langenberg C, Frants R, Willems-Vandijk K, Oostra BA, Willems SM, Lamina C, Winkler TW, Psaty BM, Tracy RP, Brody J, Chen I, Viikari J, Kähönen M, Pramstaller PP, Evans DM, St Pourcain B, Sattar N, Wood AR, Bandinelli S, Carlson OD, Egan JM, Böhringer S, van Heemst D, Kedenko L, Kristiansson K, Nuotio ML, Loo BM, Harris T, Garcia M, Kanaya A, Haun M, Klopp N, Wichmann HE, Deloukas P, Katsareli E, Couper DJ, Duncan BB, Kloppenburg M, Adair LS, Borja JB, Wilson JG, Musani S, Guo X, Johnson T, Semple R, Teslovich TM, Allison MA, Redline S, Buxbaum SG, Mohlke KL, Meulenbelt I, Ballantyne CM, Dedoussis GV, Hu FB, Liu Y, Paulweber B, Spector TD, Slagboom PE, Ferrucci L, Jula A, Perola M, Raitakari O, Florez JC, Salomaa V, Eriksson JG, Frayling TM, Hicks AA, Lehtimäki T, Smith GD, Siscovick DS, Kronenberg F, van Duijn C, Loos RJ, Waterworth DM, Meigs JB, Dupuis J, Richards JB, Voight BF, Scott LJ, Steinthorsdottir V, Dina C, Welch RP, Zeggini E, Huth C, Aulchenko YS, Thorleifsson G, McCulloch LJ, Ferreira T, Grallert H, Amin N, Wu G, Willer CJ, Raychaudhuri S, McCarroll SA, Hofmann OM, Segrè AV, van Hoek M, Navarro P, Ardlie K, Balkau B, Benediktsson R, Bennett AJ, Blagieva R, Boerwinkle E, Bonnycastle LL, Boström KB, Bravenboer B, Bumpstead S, Burtt NP, Charpentier G, Chines PS, Cornelis M, Crawford G, Doney AS, Elliott KS, Elliott AL, Erdos MR, Fox CS, Franklin CS, Ganser M, Gieger C, Grarup N, Green T, Griffin S, Groves CJ, Guiducci C, Hadjadj S, Hassanali N, Herder C, Isomaa B, Jackson AU, Johnson PR, Jørgensen T, Kao WH, Kong A, Kraft P, Kuusisto J, Lauritzen T, Li M, Lieverse A, Lindgren CM, Lyssenko V, Marre M, Meitinger T, Midthjell K, Morken MA, Narisu N, Nilsson P, Owen KR, Payne F, Petersen AK, Platou C, Proença C, Prokopenko I, Rathmann W, Rayner NW, Robertson NR, Rocheleau G, Roden M, Sampson MJ, Saxena R, Shields BM, Shrader P, Sigurdsson G, Sparsø T, Strassburger K, Stringham HM, Sun Q, Swift AJ, Thorand B, Tichet J, Tuomi T, van Dam RM, van Haeften TW, van Herpt T, van Vliet-Ostaptchouk JV, Walters GB, Weedon MN, Wijmenga C, Witteman J, Bergman RN, Cauchi S, Collins FS, Gloyn AL, Gyllensten U, Hansen T, Hide WA, Hitman GA, Hofman A, Hunter DJ, Hveem K, Laakso M, Morris AD, Palmer CN, Rudan I, Sijbrands E, Stein LD, Tuomilehto J, Uitterlinden A, Walker M, Watanabe RM, Abecasis GR, Boehm BO, Campbell H, Daly MJ, Hattersley AT, Pedersen O, Barroso I, Groop L, Sladek R, Thorsteinsdottir U, Wilson JF, Illig T, Froguel P, van Duijn CM, Stefansson K, Altshuler D, Boehnke M, McCarthy MI, Soranzo N, Wheeler E, Glazer NL, Bouatia-Naji N, Mägi R, Randall J, Elliott P, Rybin D, Dehghan A, Hottenga JJ, Song K, Goel A, Lajunen T, Doney A, Cavalcanti-Proença C, Kumari M, Timpson NJ, Zabena C, Ingelsson E, An P, O’Connell J, Luan J, Elliott A, McCarroll SA, Roccasecca RM, Pattou F, Sethupathy P, Ariyurek Y, Barter P, Beilby JP, Ben-Shlomo Y, Bergmann S, Bochud M, Bonnefond A, Borch-Johnsen K, Böttcher Y, Brunner E, Bumpstead SJ, Chen YD, Chines P, Clarke R, Coin LJ, Cooper MN, Crisponi L, Day IN, de Geus EJ, Delplanque J, Fedson AC, Fischer-Rosinsky A, Forouhi NG, Franzosi MG, Galan P, Goodarzi MO, Graessler J, Grundy S, Gwilliam R, Hallmans G, Hammond N, Han X, Hartikainen AL, Hayward C, Heath SC, Hercberg S, Hillman DR, Hingorani AD, Hui J, Hung J, Kaakinen M, Kaprio J, Kesaniemi YA, Kivimaki M, Knight B, Koskinen S, Kovacs P, Kyvik KO, Lathrop GM, Lawlor DA, Le Bacquer O, Lecoeur C, Li Y, Mahley R, Mangino M, Martínez-Larrad MT, McAteer JB, McPherson R, Meisinger C, Melzer D, Meyre D, Mitchell BD, Mukherjee S, Naitza S, Neville MJ, Orrù M, Pakyz R, Paolisso G, Pattaro C, Pearson D, Peden JF, Pedersen NL, Pfeiffer AF, Pichler I, Polasek O, Posthuma D, Potter SC, Pouta A, Province MA, Rayner NW, Rice K, Ripatti S, Rivadeneira F, Rolandsson O, Sandbaek A, Sandhu M, Sanna S, Sayer AA, Scheet P, Seedorf U, Sharp SJ, Shields B, Sigurðsson G, Sijbrands EJ, Silveira A, Simpson L, Singleton A, Smith NL, Sovio U, Swift A, Syddall H, Syvänen AC, Tönjes A, Uitterlinden AG, van Dijk KW, Varma D, Visvikis-Siest S, Vitart V, Vogelzangs N, Waeber G, Wagner PJ, Walley A, Ward KL, Watkins H, Wild SH, Willemsen G, Witteman JC, Yarnell JW, Zelenika D, Zethelius B, Zhai G, Zhao JH, Zillikens MC, Borecki IB, Meneton P, Magnusson PK, Nathan DM, Williams GH, Silander K, Bornstein SR, Schwarz P, Spranger J, Karpe F, Shuldiner AR, Cooper C, Serrano-Ríos M, Lind L, Palmer LJ, Hu FB, Franks PW, Ebrahim S, Marmot M, Kao WH, Pramstaller PP, Wright AF, Stumvoll M, Hamsten A, Buchanan TA, Valle TT, Rotter JI, Penninx BW, Boomsma DI, Cao A, Scuteri A, Schlessinger D, Uda M, Ruokonen A, Jarvelin MR, Peltonen L, Mooser V, Sladek R, Musunuru K, Smith AV, Edmondson AC, Stylianou IM, Koseki M, Pirruccello JP, Chasman DI, Johansen CT, Fouchier SW, Peloso GM, Barbalic M, Ricketts SL, Bis JC, Feitosa MF, Orho-Melander M, Melander O, Li X, Li M, Cho YS, Go MJ, Kim YJ, Lee JY, Park T, Kim K, Sim X, Ong RT, Croteau-Chonka DC, Lange LA, Smith JD, Ziegler A, Zhang W, Zee RY, Whitfield JB, Thompson JR, Surakka I, Spector TD, Smit JH, Sinisalo J, Scott J, Saharinen J, Sabatti C, Rose LM, Roberts R, Rieder M, Parker AN, Pare G, O’Donnell CJ, Nieminen MS, Nickerson DA, Montgomery GW, McArdle W, Masson D, Martin NG, Marroni F, Lucas G, Luben R, Lokki ML, Lettre G, Launer LJ, Lakatta EG, Laaksonen R, Kyvik KO, König IR, Khaw KT, Kaplan LM, Johansson Å, Janssens AC, Igl W, Hovingh GK, Hengstenberg C, Havulinna AS, Hastie ND, Harris TB, Haritunians T, Hall AS, Groop LC, Gonzalez E, Freimer NB, Erdmann J, Ejebe KG, Döring A, Dominiczak AF, Demissie S, Deloukas P, de Faire U, Crawford G, Chen YD, Caulfield MJ, Boekholdt SM, Assimes TL, Quertermous T, Seielstad M, Wong TY, Tai ES, Feranil AB, Kuzawa CW, Taylor HA, Gabriel SB, Holm H, Gudnason V, Krauss RM, Ordovas JM, Munroe PB, Kooner JS, Tall AR, Hegele RA, Kastelein JJ, Schadt EE, Strachan DP, Reilly MP, Samani NJ, Schunkert H, Cupples LA, Sandhu MS, Ridker PM, Rader DJ, Kathiresan S. Novel loci for adiponectin levels and their influence on type 2 diabetes and metabolic traits: a multi-ethnic meta-analysis of 45,891 individuals. PLoS Genet 2012;8:e1002607. https://doi.org/10.1371/journal.pgen.1002607
- 34. Breitfeld J, Stumvoll M, Kovacs P. Genetics of adiponectin. Biochimie 2012;94:2157-2163. https://doi.org/10.1016/j.biochi.2012.03.004
- 35. Balatskiy A, Teterina M, Pisaryuk A, Balabanenko I, Kadrev A, Tishuk A, Balatskaya M, Samokhodskaya L, Boytsov S, Kalinina N, Tkachuk V. T-cadherin and the ratio of its ligands as predictors of carotid atherosclerosis: a pilot study. Biomedicines 2021;9:1398. https://doi.org/10.3390/biomedicines9101398
- 36. Parker-Duffen JL, Nakamura K, Silver M, Kikuchi R, Tigges U, Yoshida S, Denzel MS, Ranscht B, Walsh K. T-cadherin is essential for adiponectin-mediated revascularization. J Biol Chem 2013;288:24886-24897. https://doi.org/10.1074/jbc.M113.454835
- 37. Becic T, Studenik C, Hoffmann G. Exercise increases adiponectin and reduces leptin levels in prediabetic and diabetic individuals: systematic review and meta-analysis of randomized controlled trials. Med Sci (Basel) 2018;6:97. https://doi.org/10.3390/medsci6040097
- 38. Maldonado M, Chen J, Duan H, Zhou S, Yang L, Raja MA, Huang T, Jiang G, Zhong Y. Effects of caloric overload before caloric restriction in the murine heart. Aging (Albany NY) 2022;14:2695-2719. https://doi.org/10.18632/aging.203967
- 39. Wu P, Wen W, Li J, Xu J, Zhao M, Chen H, Sun J. Systematic review and meta-analysis of randomized controlled trials on the effect of SGLT2 inhibitor on blood leptin and adiponectin level in patients with type 2 diabetes. Horm Metab Res 2019;51:487-494. https://doi.org/10.1055/a-0958-2441
- 40. Sugiyama S, Fukushima H, Kugiyama K, Maruyoshi H, Kojima S, Funahashi T, Sakamoto T, Horibata Y, Watanabe K, Koga H, Sugamura K, Otsuka F, Shimomura I, Ogawa H. Pravastatin improved glucose metabolism associated with increasing plasma adiponectin in patients with impaired glucose tolerance and coronary artery disease. Atherosclerosis 2007;194:e43-e51. https://doi.org/10.1016/j.atherosclerosis.2006.08.023
- 41. Gianopoulos I, Mantzoros CS, Daskalopoulou SS. Adiponectin and adiponectin receptors in atherosclerosis. Endocr Rev 2025;46:1-25. https://doi.org/10.1210/endrev/bnae021
- 42. Fujii K, Fujishima Y, Kita S, Kawada K, Fukuoka K, Sakaue TA, Okita T, Kawada-Horitani E, Nagao H, Fukuda S, Maeda N, Nishizawa H, Shimomura I. Pharmacological HIF-1 activation upregulates extracellular vesicle production synergistically with adiponectin through transcriptional induction and protein stabilization of T-cadherin. Sci Rep 2024;14:3620. https://doi.org/10.1038/s41598-024-51935-6