1Department of Internal Medicine, Chung-Ang University College of Medicine, Seoul, Korea
*Corresponding author: Han Ah Lee,
Department of Internal Medicine, Chung-Ang University College of Medicine, 102,
Heukseok-ro, Dongjak-gu, Seoul 06973, Korea, E-mail:
amelia86@naver.com
• Received: August 29, 2024 • Accepted: September 29, 2024
This is an Open-Access article distributed under the terms of the
Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0) which permits
unrestricted non-commercial use, distribution, and reproduction in any
medium, provided the original work is properly cited.
Metabolic dysfunction-associated steatohepatitis (MASH) is increasingly
recognized as a leading cause of hepatocellular carcinoma (HCC), the
third-leading cause of cancer mortality worldwide, driven by the global obesity
epidemic. Projected to become the primary cause of HCC by 2030, MASH-HCC
presents unique clinical challenges. This review examines its clinical
management, including surveillance strategies and treatment advances, and
discusses prospects to overcome existing challenges. MASH-HCC accounts for
10%–20% of HCC cases, particularly in Western countries, with a rising
incidence due to obesity. Risk factors include cirrhosis, diabetes, obesity,
alcohol, smoking, genetic polymorphisms (e.g., PNPLA3), and microbiome
alterations. The pathogenesis involves fibrosis, immune dysfunction (e.g.,
T-cell impairment), and molecular changes. Prevention focuses on lifestyle
modifications. Surveillance in patients with MASH cirrhosis is crucial but is
hindered by poor ultrasound sensitivity in obese patients, necessitating
alternative methods. Treatment mirrors that of other HCC types, but
comorbidities and potentially reduced efficacy of immunotherapy necessitate
tailored approaches. MASH is becoming the leading cause of HCC, necessitating
lifestyle interventions for prevention. Improved surveillance and early
detection are critical but challenging due to obesity-related factors.
Treatments align with those for other HCC types, but comorbidities and potential
differences in immunotherapy efficacy due to T-cell dysfunction require careful
consideration. Key needs include identifying molecular drivers in non-cirrhotic
metabolic dysfunction-associated steatotic liver disease, developing preventive
therapies, refining surveillance methods, and tailoring treatments. Trials
should specifically report MASH-HCC outcomes to enable personalized therapies.
Further research is needed to understand T-cell dysfunction, optimize
immunotherapies, and identify predictive biomarkers.
Primary liver cancer ranks as the third leading cause of cancer-related deaths
globally, with nearly 1 million new cases reported annually [1]. Hepatocellular carcinoma (HCC) accounts
for approximately 90% of all primary liver cancers. It typically arises in the
setting of chronic liver diseases, which may be due to HBV, HCV, alcohol-related
liver disease, or metabolic dysfunction-associated steatotic liver disease
(MASLD) [2]. MASLD is estimated to affect
around 20%–25% of the global population [3]. Metabolic dysfunction-associated steatohepatitis (MASH) is
characterized by more than 5% steatosis, hepatocellular injury (such as
"ballooning"), and inflammation, which may occur with or without
fibrosis [4]. About 20% of individuals
with MASLD develop MASH, which is strongly linked to rising rates of obesity,
diabetes, and metabolic syndrome. As MASH progresses, it can lead to severe
liver-related complications, including cirrhosis or liver failure, and
significantly increases the risk of developing HCC [5].
In patients with MASH-related cirrhosis, the annual incidence of HCC is
approximately 2% [6]. Moreover, MASH is
the primary cause of HCC in patients who do not have cirrhosis [7]. MASH-related HCC accounts for 20% of HCC
cases in the Western world and is projected to become the leading cause of HCC
globally by 2030 [8]. The development of
MASH-related HCC is characterized by unique mutational, immunological, and
microenvironmental features. Although most cases of MASH-related HCC occur in
patients with cirrhosis, 30%–40% develop in those with advanced fibrosis
but without cirrhosis. This suggests a distinct metabolic environment and the
likely involvement of extrahepatic cancer drivers associated with metabolic
syndrome [9,10]. Unlike infections with HBV or HCV, MASH more
frequently leads to HCC in the absence of cirrhosis, underscoring the need for
strengthened surveillance and early detection [11].
Currently, MASH-HCC is managed similarly to other causes of HCC, employing
strategies such as transplantation, resection, or locoregional therapies for
early- or intermediate-stage disease [12]. MASH is the leading cause of HCC-related liver transplants in the
USA; however, approximately 50% of patients undergo systemic therapy as their
disease progresses, which includes both combination therapies and single-agent
treatments with tyrosine kinase inhibitors or monoclonal antibodies [13]. Nevertheless, it remains uncertain
whether immune-based therapies are as effective for non-viral HCC as they are
for viral-related HCC [14].
Objectives
In this review, we examine the clinical management of MASH-HCC, focusing on
surveillance strategies and recent advancements in treatment. We also discuss
the customized application and outcomes of surgical, locoregional, and systemic
therapies, examining future prospects and strategies to address current
challenges.
Ethics statement
It is a literature database-based review; therefore, neither approval by the
institutional review board nor obtainment of informed consent was required.
Epidemiology
Approximately 10% (ranging from 1% to 38%) of all HCC cases are associated with
MASLD, with higher rates (>20%) reported in studies from the USA, UK, India,
Germany, and the Middle East. In contrast, lower estimates (1%–2%) are
reported from China and Japan [15]. The
incidence of MASH-related HCC is expected to rise substantially as the obesity
epidemic continues to expand [16].
Mathematical models predict a significant increase in the incidence of MASH-HCC from
2016 to 2030, with projected rises of 47% in Japan, 82% in China, 88% in the UK,
117% in France, and 130% in the USA [17].
Compared to patients with HCC due to viral hepatitis (HBV or HCV) or alcohol-related
liver disease, those with MASH-HCC typically have a lower male-to-female ratio
(1.2:1), are generally 5–10 years older (mean age 73), and are more likely to
have metabolic and cardiovascular comorbidities, such as type 2 diabetes mellitus
(DM) and chronic vascular disease. Although the incidence of MASH-HCC is lower than
that associated with active viral hepatitis, the increasing prevalence of MASLD,
combined with improved treatments for viral hepatitis, is expected to increase both
the proportion and rate of HCC attributed to MASLD [18,19].
Risk factors
Liver cirrhosis
A study involving approximately 300,000 patients with MASLD reported an HCC
incidence of 0.21 per 1,000 person-years, which is seven times higher than that
observed in control individuals without liver disease—specifically, those
free from viral hepatitis and with normal alanine aminotransferase levels [20]. The primary risk factor for MASH-HCC
is cirrhosis, with incidence rates in cohorts of MASH cirrhosis estimated at
about 2% per year, although these rates vary from 0.3% to 4.7% per year [6]. This variability can be attributed to
differences in age, metabolic profiles, and the severity of liver
decompensation. While HCC can also develop in MASH patients without cirrhosis,
the overall incidence in this subgroup is low, ranging between 0.01% and 0.13%
per year. It is even lower in the general MASLD population, underscoring the
importance of assessing cirrhosis status as the primary risk stratifier for
MASLD [21].
Diabetes
In cohort studies from both Europe and the US, type 2 diabetes has been
identified as the strongest independent metabolic risk factor for the
development of HCC. A retrospective study demonstrated that in patients with
MASH-cirrhosis, the presence of DM was associated with a fourfold increase in
the risk of developing HCC (hazard ratio [HR], 4.2; 95% CI, 1.2–14.2;
P=0.02) [19]. Another large study in
Europe, which included 136,703 patients with MASLD, found that among the 6,425
(4.7%) patients with advanced fibrosis, DM was the most significant risk factor
for HCC [22]. Similarly, a study
involving a US cohort of 271,906 MASLD patients, of whom 253 had HCC, reported a
strong association between DM and HCC (adjusted HR, 2.77; 95% CI,
2.03–3.77) [23].
Obesity
In a large cohort study involving 296,707 patients, those diagnosed with MASLD
and obesity did not show a statistically significant increase in HCC risk
(P=0.06). However, the risk increased significantly, by 2.6 times, when obesity
was accompanied by diabetes, hypertension, and hyperlipidemia [20]. Another recent study, which examined
data from 98,090 MASLD patients with severe obesity, found that those who
underwent bariatric surgery experienced a reduced risk of HCC. The adjusted HR
was 0.48 (95% CI, 0.24–0.89) [24].
Although numerous studies have explored the link between obesity and elevated
HCC risk, most have not sufficiently evaluated the presence of MASLD or
MASH.
Alcohol
The impact of mild to moderate alcohol consumption on the development of HCC in
patients with MASLD is still unclear, as research has produced inconsistent
findings. A cohort study in Korea examined the relationship between mild to
moderate alcohol intake and the progression of non-invasive fibrosis scores in
58,927 adults with MASLD who initially had low fibrosis scores over a median
period of 4.9 years [25]. Of these
participants, 5,303 (9%) progressed from low to intermediate or high fibrosis
scores. Moderate drinkers were more likely to experience increased fibrosis
compared to nondrinkers, with an HR of 1.29 (95% CI, 1.23). Another study
indicated that even mild drinking habits increased the risk of carcinogenesis in
patients with MASH-associated cirrhosis, presenting an HR of 3.8 (95% CI,
1.6–8.9; P=0.002); however, this study focused solely on patients with
decompensated liver disease [26].
Additionally, a recent multivariate analysis of patients with biopsy-proven
MASLD across various stages of fibrosis revealed that consuming less than
20 g of alcohol per day heightened the risk of HCC, especially in those
with advanced F3–4 fibrosis, with a relative risk of 4.83 (P=0.04) [27].
Smoking
Smoking is generally associated with an increased risk of HCC; however, its
specific impact on MASLD has not been thoroughly investigated [28].
Coffee
Coffee is rich in antioxidants, including phenolic compounds such as chlorogenic,
caffeic, ferulic, and coumaric acids, along with melanoidins and diterpenes such
as cafestol and kahweol. These compounds have shown inhibitory effects on the
development of HCC [29,30]. Additionally, the beneficial effects
of coffee in preventing HCC may be partially attributed to its role in lowering
the risk of type 2 DM, which is a known risk factor for HCC [31].
Antidiabetics
Metformin inhibits the mammalian target of the rapamycin pathway, which plays a
role in cell proliferation by activating AMP-activated protein kinase (AMPK)
[32]. It also inhibits angiogenesis,
disrupts the cell cycle, and induces apoptosis independently of p53 [33]. Additionally, metformin promotes
moderate weight loss, mitigates the effects of hyperinsulinemia on the cell
cycle and inflammation, and improves liver biochemistry and histology in
patients with MASLD [34,35]. Research has explored the impact of
antidiabetic medications on HCC risk, recognizing diabetes as a significant risk
factor. A recent study demonstrated that effective glycemic control was
associated with a 31% reduced risk of HCC in patients with MASLD and DM [36]. The study also found that metformin
use led to a 20% decrease in HCC risk, whereas insulin use, particularly when
combined with other oral antidiabetic medications, increased the risk by 1.6 to
1.7 times. However, a database study of 18,080 MASLD patients without cirrhosis,
monitored over an average of 6.3 years, showed no link between metformin use and
HCC risk [37]. In a recent nationwide
cohort, patients with MASLD and DM who used sodium-glucose cotransporter-2
inhibitors had significantly lower risks of liver and non-liver complications
compared to users of other antidiabetic medications, with HRs ranging from 0.76
to 0.97. The risk was further reduced when metformin was also used, with HRs
between 0.58 and 0.79 [38].
Statins
Statins exhibit a range of anticancer effects that go beyond their ability to
lower cholesterol. They inhibit key oncogenic drivers including MYC, AKT,
Rho-dependent kinase, and extracellular signal-regulated kinase 1 and 2 [35,39,40]. Additionally, statins
activate protective liver pathways such as AMPK and p38-MAPK, and promote
apoptosis through a p53-dependent mechanism [41,42]. These drugs have also
been linked to anticarcinogenic effects. A database study from Taiwan involving
18,080 MASLD patients demonstrated an inverse relationship between statin use
and HCC, with an OR of 0.29 (95% CI, 0.12–0.68) [37]. In a retrospective case-control study of 102 MASLD
patients, including 34 HCC cases, statins were found to be protective against
HCC (OR, 0.20; 95% CI, 0.07–0.60) [43]. Another recent retrospective study showed that statin use
significantly and dose-dependently reduced the risk of HCC in patients with NASH
cirrhosis [44]. However, a study
involving 458 MASLD patients with advanced fibrosis did not find such an
association [45]. The uncontrolled and
retrospective nature of these studies limits the ability to definitively
interpret their findings on the chemopreventive benefits of statins, making it
inappropriate to recommend them solely for the prevention of HCC.
Pathogenesis
Liver fibrosis
Approximately 80% of MASLD patients do not develop NASH, prompting research
efforts to focus on identifying the factors that differentiate those with
inflammation, cell injury, and fibrosis (MASH) from those exhibiting simple
steatosis. A critical factor in understanding the progression to MASH is
lipotoxicity, which involves hepatocellular injury resulting from disrupted fat
metabolism [46]. Lipotoxicity is
triggered by various factors, including increased fatty acid delivery to the
liver, insulin resistance, and inflammatory signals from dysfunctional adipose
tissue [47]. This condition leads to
cellular stress, oxidative damage, inflammasome activation, and ultimately, cell
death in hepatocytes [48]. These damaging
responses are linked to pre-malignant changes, such as oxidative DNA damage and
mutations in metabolism-related genes such as FOXO1, CIDEB, and
GPAM. Although these genes may help protect hepatocytes
from lipotoxicity, they also elevate the risk of malignancy [49,50].
To repair hepatocellular injuries in MASH, developmental pathways such as
YAP–TAZ, Notch, and Hedgehog signaling are reactivated in hepatocytes.
This reactivation leads to cell proliferation, inflammation, and potentially
cancer [51,52]. In advanced MASH, there is a marked decline in
hepatocyte proliferation and regenerative capacity. These dysregulated cells
exacerbate inflammation and fibrosis [53]. Consequently, this hepatocellular damage fosters a pro-inflammatory
environment, perpetuating chronic inflammation and impacting various immune cell
types.
The stage of hepatic fibrosis in MASH is a critical determinant of clinical
outcomes, as it can progress to cirrhosis and liver failure, and create
conditions conducive to cancer development [54]. This process involves the activation or transdifferentiation of
resident hepatic stellate cells (HSCs) into fibrogenic, proliferating
myofibroblasts, which leads to the accumulation of extracellular matrix or scar
tissue. Advanced single-cell sequencing has revealed significant heterogeneity
among HSCs in MASH, although the functional implications of this diversity are
not yet clear [55]. The exact mechanisms
by which MASH-HCC develops without cirrhosis remain poorly understood, but they
are likely related to fibrosis. The accumulation of extracellular matrix
increases liver stiffness, which can facilitate the emergence and growth of
tumor cells [56]. This scar matrix also
acts as a reservoir for growth factors that may support the survival of
pre-neoplastic hepatocytes, thereby promoting tumor initiation or progression.
Additionally, HSCs possess immunoregulatory properties that contribute to the
liver's immune tolerance, potentially affecting its response to
checkpoint blockade therapies [57].
Angiogenesis is implicated in both MASH and potentially MASH-HCC. Increased CD34
expression in new blood vessels has been observed in previous studies involving
both humans and rodents, indicating enhanced vascularization [58]. Vascular endothelial growth factor
(VEGF), a crucial angiogenic signal, shows elevated levels in experimental MASH
models. Inhibiting VEGF leads to reduced vascularization, inflammation, and
steatosis [59].
The impact of treatments targeting MASH on the risk of MASH-HCC has yet to be
determined; however, a decrease in HCC risk has been noted in MASH patients
following bariatric surgery, indicating that future medical interventions for
MASH could potentially lower the incidence of HCC [60]. Nonetheless, it remains uncertain whether advanced
liver fibrosis continues to carry an inherent risk of cancer even if the
fibrosis subsequently regresses.
Immune system
The immune system plays a major role in both MASLD and HCC, and distinct
immunogenomic classifications have been identified [61]. MASH is characterized by inflammatory responses in the
liver, which are pivotal in its progression to fibrosis, cirrhosis, or HCC
[62]. Both innate and adaptive immune
mechanisms significantly contribute to hepatic inflammation in MASH. Resident
Kupffer cells and the recruitment of leukocytes, including neutrophils,
monocytes, NK cells, and NKT cells, promote inflammation through the release of
cytokines, chemokines, and reactive oxygen species. Elevated levels of CD4+ T
helper cells, particularly the TH1 and TH17 subsets, have been observed in the
livers of mice with MASH [63]. Although T
cells exhibit anti-tumorigenic properties, the depletion of CD8+ T cells
accelerates tumor growth in MASH-driven HCC models. Similarly, the depletion of
CD4+ T cells promotes tumor growth, impacting the efficacy of immune-based
therapies [64].
The disruption of the immune system in MASH and MASH-HCC has been linked to the
response to immunotherapies. Both adaptive and innate immune cells, including
CD4+ T cells, metabolically activated CD8+ T cells, platelets, and dendritic
cells, play a role in shaping the liver microenvironment as MASH progresses to
HCC [65,66]. Neutrophils, in particular, are involved in the transition from
fatty liver to steatohepatitis. They contribute to an immunosuppressive
environment through the production of extracellular traps and PDL1 signaling,
which leads to CD8+ T cell exhaustion and affects the response to immunotherapy
[67,68]. In MASLD, impaired antigen-specific T-cell function has been
observed, partially due to macrophage activity [69]. In advanced HCC, the infiltration of CCR2+ and CX3CR1+
macrophages is linked to non-responsiveness to immune-checkpoint inhibition.
Conversely, pro-inflammatory PDL1-expressing CXCL10+ macrophages can drive
treatment response. Recent studies indicate that T cells lose functionality in
MASLD, which contributes to poor responses to immune checkpoint inhibitor (ICI)
therapy [70]. Approaches such as
neutrophil reprogramming with CXCR2 antagonists have shown promise in enhancing
the effectiveness of ICI therapy in MASH-HCC models by increasing dendritic cell
activity and CD8+ T cell numbers [68].
In two notable studies involving both mice and humans, the presence of CD8+PD1+ T
cells in the liver increased as MASH progressed. These cells are in an
auto-aggressive state, characterized by liver-resident CD8+PD1+CD103+ T cells
that, despite being exhausted, display an activated phenotype and express high
levels of cytokines such as TNF, CCL2, IL-10, and granzyme B [71,72]. In MASH-HCC mouse models treated with immunotherapy, these
CD8+PD1+ cells exhibited minimal changes in their transcriptomes and proteomes,
yet they increased in size over time. This growth contributed to heightened
liver inflammation, hepatocyte death, and oncogenic signaling [72]. Instead of eliminating HCC, these
cells became dysfunctional in tumor surveillance and even promoted tumor growth.
This dysfunction resulted in a lack of response to ICIs in therapeutic settings
and accelerated HCC development in preventive scenarios. Similar characteristics
of CD8+ T cells have been observed in human MASH-HCC, indicating that
peritumoral and intratumoral CD8+PD1+ T cells could potentially serve as
predictors of treatment success or resistance to ICIs. Understanding the immune
microenvironment is essential for identifying the most effective therapies in
future research.
Microbiome
The gut microbiome plays a crucial role in influencing altered liver responses in
MASH by affecting hepatic bile acid metabolism and facilitating the
translocation of gut-derived signals through an increasingly permeable gut
lining [73]. Throughout all stages of
NASH, the gut–liver axis remains active, with interactions between liver
damage, regeneration, and heightened gut permeability exacerbating inflammatory,
pro-fibrogenic, and pro-carcinogenic pathways [48]. This permeability defect allows for both direct (e.g.,
bacterial presence) and indirect (e.g., bacterial metabolites) interactions
between the gut microbiome and the liver, which in turn impact liver metabolism
and contribute to the progression of MASH and HCC.
The gut microbiome has been identified as a key factor in triggering MASLD,
driving liver steatosis by enhancing energy harvest, monosaccharide absorption,
and abnormal acetate production [74]. A
dysbiotic, leaky gut permits the translocation of pathogen-associated and
danger-associated molecular patterns into the liver, activating immune cells and
Toll-like receptors, which in turn trigger pro-inflammatory and fibrotic
pathways [75]. In mice, disruption of the
gut vascular barrier by the microbiota is seen as a precursor to NASH [76]. Additionally, inflammatory cells from
the gut may migrate to the liver, contributing to bacterial translocation.
Several bacterial species, such as Proteobacteria, Enterobacteriaceae, and
Escherichia, are associated with MASLD in humans, and levels of Bacteroides are
elevated in MASH patients [76,77]. Treatment with non-absorbable
antibiotics, such as rifaximin, has shown potential in improving liver function,
underscoring the significant role of the gut microbiome in MASH pathogenesis
[78].
Molecular alterations
Several single-nucleotide polymorphisms (SNPs) associated with abnormal lipid
metabolism in hepatocytes have been linked to an increased risk of MASH and
progression to HCC. One of the most well-known SNPs is rs738409 in the
PNPLA3 gene, which encodes the patatin-like phospholipase
domain-containing protein 3. This variant interferes with the breakdown of lipid
droplets in hepatocytes, leading to decreased triglyceride lipolysis and
promoting hepatic steatosis. As a result, it is associated with more than a
2-fold increased risk of MASH and a 2.2-fold higher risk of progressing to MASH
HCC compared to those without the variant [79]. Another significant SNP, rs58542926 in the
TM6SF2 gene, plays a role in regulating liver fat
metabolism and increases hepatic triglyceride content. This variant is linked to
a 1.6-fold increased risk of MASH and a 1.9-fold higher risk of MASH HCC [80].
Additionally, an SNP near the MBOAT7 gene is associated with
increased hepatic triglyceride levels and occurs twice as frequently in patients
with MASH-HCC compared to those with MASLD alone [81]. A loss-of-function variant in the GCKR gene, which
encodes the glucokinase regulator, leads to increased de novo
lipogenesis and insulin resistance. This variant is linked to a 1.5-fold
increased risk of MASH and a 1.8-fold higher risk of MASH-HCC [82]. A polygenic risk score that
incorporates these four SNPs has been suggested for HCC risk stratification in
patients of European ancestry with NASH cirrhosis. This score has proven to be a
more accurate predictor of HCC development than individual SNPs
(P<10–13) [83].
MASH-HCC is often associated with an increased presence of ACVR2A and TP53
mutations, as well as the proliferative class S1-WNT/TGFβ [84]. A distinct mutational signature,
termed MutSigNASH-HCC, has been identified in 25% of MASH-HCC patients, compared
to only 2% in those with other causes. This signature is characterized by a
higher frequency of C>T and C>A transitions [85]. Furthermore, patients with MASH-HCC exhibit higher
levels of hepatic oxidative DNA damage than those with other etiologies, a
phenomenon that correlates with a diminished DNA damage response in experimental
models [49]. Additionally, epigenetic
events that suppress the transcription of genes involved in bile and fatty acid
metabolism, while activating proliferative pathways, have been implicated in
MASH-HCC. Experimental models have shown that epigenetic reprogramming can
reverse hepatocarcinogenesis [86].
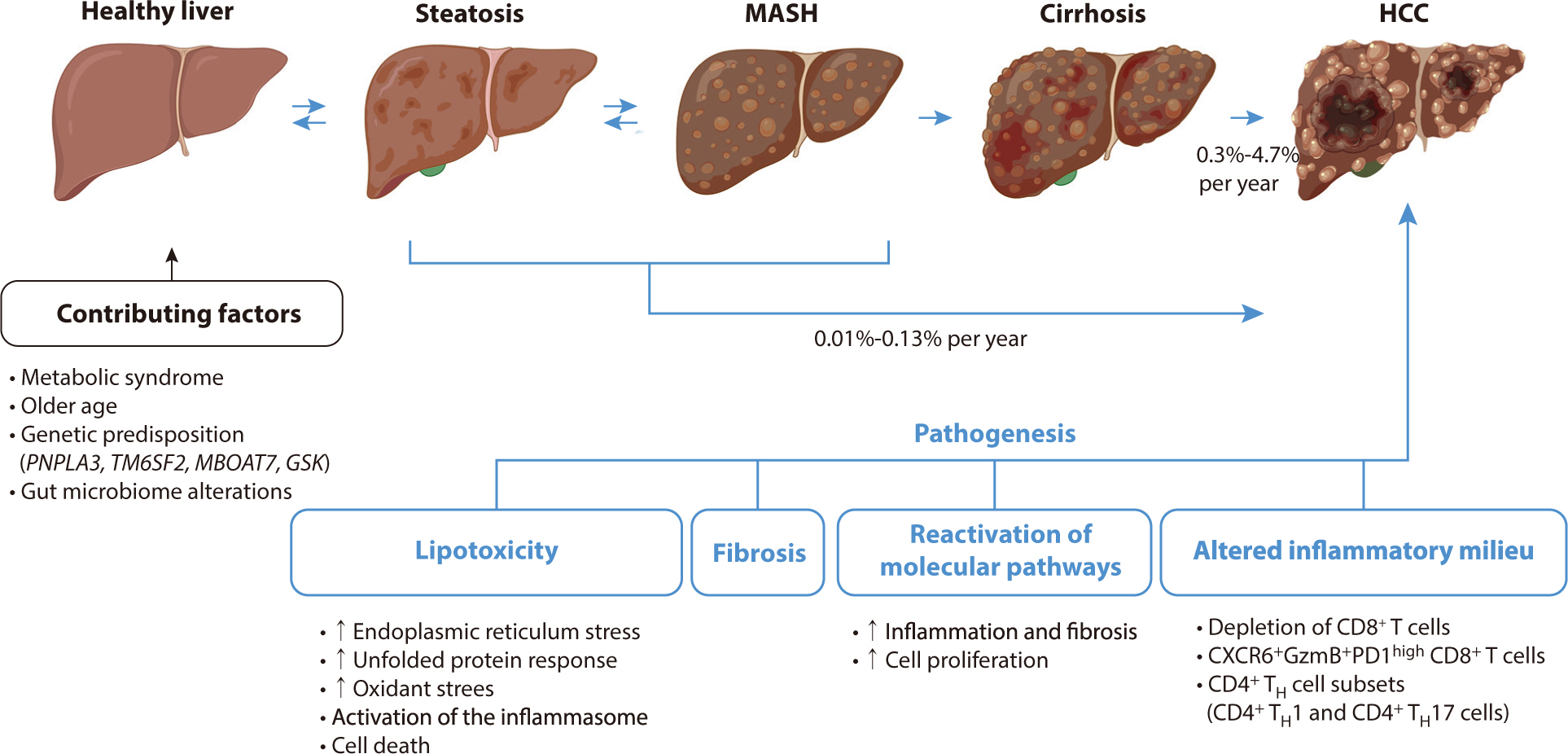
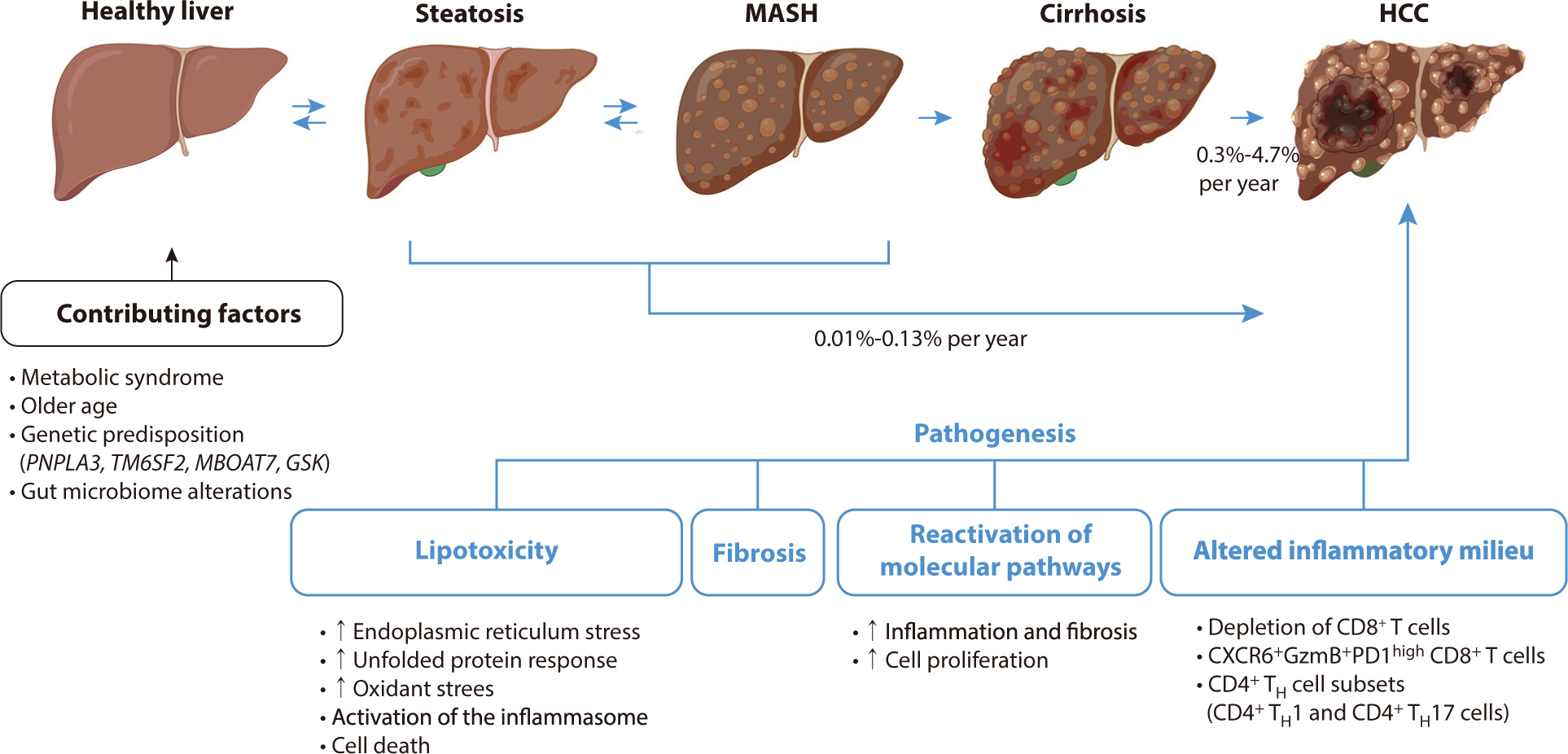
A diagram of the pathogenesis of HCC associated with metabolic
dysfunction-related steatohepatitis is shown in Fig. 1.
Fig. 1.
Pathogenesis and progression of MASH-HCC (drawn by the author). MASH,
metabolic dysfunction-associated steatohepatitis; HCC, hepatocellular
carcinoma; TH, T helper.
Clinical management
Prevention
Several observational, retrospective, population-based studies have suggested
that metformin, statins, coffee, and aspirin might contribute to the prevention
of HCC, regardless of the underlying liver disease etiology [87,88]. Due to its generally favorable benefit-to-risk ratio, current
guidelines endorse the consumption of coffee for individuals with chronic liver
disease [89,90]. However, other agents have not demonstrated sufficient
efficacy to be recommended for HCC prevention, and most studies related to this
have not been conducted in well-defined populations with MASLD.
For the prevention of MASH-HCC, the American Association for the Study of Liver
Diseases (AASLD), the European Association for the Study of the Liver (EASL),
and the Korean Association for the Study of Liver Diseases (KASL) recommend
combining a hypocaloric or Mediterranean diet with moderate-intensity exercise
to achieve and maintain weight loss, as outlined in their practice guidelines
[89–91]. Additionally, a large multinational cohort study has
demonstrated that physical activity is associated with a reduced risk of HCC
[92]. Although there is no direct
evidence currently available that weight loss decreases the risk of MASH-HCC,
observational studies indicate that weight loss may reverse steatosis and
potentially fibrosis in patients with MASH, thereby suggesting a possible
benefit of weight loss in reducing the risk of HCC [93,94].
Hepatocellular carcinoma surveillance
The clinical practice guidelines from the AASLD, EASL, and KASL recommend
semiannual surveillance for HCC using abdominal ultrasound, with or without
α-fetoprotein testing, for all patients with cirrhosis, regardless of the
underlying cause. However, only two studies have specifically assessed the
potential benefits of such surveillance in patients with MASLD-related
cirrhosis.
However, a previous study found no significant association between surveillance
and the applicability of curative treatment (45.5% versus 51.5%; P=0.72) [95].
Data specifically focusing on patients with MASLD are important, as this group
exhibits unique characteristics that pose challenges to traditional HCC
surveillance methods. Notably, about one-third of MASLD-HCC cases arise in
individuals without cirrhosis, suggesting that these patients are often excluded
from the at-risk populations typically targeted for surveillance [21]. Furthermore, at the time of their HCC
diagnosis, patients with MASLD are generally less likely to have been previously
diagnosed with liver disease or cirrhosis, which likely contributes to their
lower rates of surveillance utilization [96]. A meta-analysis revealed that a significantly smaller
proportion of patients with MASLD-HCC (32.8%, 95% CI, 12.0–63.7)
underwent surveillance compared to patients with HCC from other causes (55.7%,
95% CI, 24.0–83.3; P<0.0001) [97].
Second, patients with MASH are more likely to experience inadequate ultrasound
visualization and surveillance failure, leading to a higher rate of late-stage
HCC diagnoses even when surveillance is performed [98,99]. This
suggests that the sensitivity of ultrasound-based surveillance in patients with
MASH may be lower than the 63% observed in those with HCC from other causes
[100]. This finding underscores the
need for alternative imaging methods, such as CT or MRI, and blood-based
biomarker strategies for this group [101].
Treatment for metabolic dysfunction-associated steatohepatitis-hepatocellular
carcinoma
Patients with MASH-HCC often present with comorbidities, such as cardiovascular
disease, which can restrict their access to curative treatments, especially
surgery [102,103]. However, a systematic review has shown that despite
having more comorbidities and larger tumors at diagnosis, the allocation of
treatments for MASLD patients is similar to that for other patients [97]. Moreover, when severe comorbidities
are excluded, the outcomes following curative and locoregional treatments for
MASH-HCC are comparable to, or even better than, those observed in non-MASH
patients. Lastly, immunotherapies may be less effective in non-viral HCC cases,
such as MASH-HCC, due to impairments in the immune system [72].
Surgery: Patients with MASLD face a higher risk of intra-operative complications
and poorer post-surgical outcomes, largely due to the increased prevalence of
metabolic syndrome comorbidities. Obesity and type 2 diabetes have been linked
to lower survival rates in cancer patients, including those receiving surgical
treatments [104,105]. Research indicates that patients with MASH-HCC are
more likely to suffer from hypertension, hyperlipidemia, and ischemic heart
disease compared to those with other causes of HCC, all factors that heighten
the risk of post-surgical morbidity and complications [102]. Furthermore, the degree of liver steatosis may
correlate with poorer surgical outcomes [106].
However, a systematic review and meta-analysis of 14 studies, which included
7,226 HCC patients—approximately 20% of whom had
MASH-HCC—demonstrated that patients with MASH-HCC experienced improved
disease-free survival (HR, 0.81) and overall survival (HR, 0.78) compared to
those with other causes [107]. Another
meta-analysis corroborated these results, suggesting that the better outcomes in
MASH-HCC patients might be due to the absence of cirrhosis in many cases and the
exclusion of those with severe comorbidities from surgical interventions [108].
Liver transplantation: An analysis of the United Network for Organ Sharing (UNOS)
registry from 2002 to 2012 revealed that patients with MASH-HCC had
significantly better post-transplant survival outcomes (HR, 0.69; 95% CI,
0.63–0.77) and a lower risk of graft failure (HR, 0.76; 95% CI,
0.69–0.83) compared to those with other causes of HCC. This was despite a
higher prevalence of diabetes and cardiovascular disease in the MASH-HCC group
[109]. In contrast, data from the
European Liver Transplant Registry showed no statistically significant
differences in post-transplant survival or graft survival between patients with
HCC, regardless of MASLD status. However, there were differences in the causes
of mortality [110]. While some
single-center studies suggest that patients with MASLD may have a higher risk of
post-transplant complications, the overall evidence indicates similar
post-transplant survival rates between patients with MASLD and those with other
etiologies of HCC [111].
Locoregional therapies: Current evidence on the efficacy of locoregional
therapies for MASH-HCC is limited. However, a study using the SEER-Medicare
database showed similar overall survival rates following radiofrequency ablation
in patients with MASH-HCC compared to those with other HCC etiologies [112]. Additionally, a propensity
score-matched study that included patients undergoing transarterial
chemoembolization revealed no significant differences in time-to-progression
(13.0 vs. 8.5 months; P=0.25) or overall survival (23.2 vs. 28.0 months; P=0.48)
between patients with and without MASLD [113]. Another study comparing MASLD-HCC and HBV-related HCC patients
treated with transarterial radioembolization also found no significant
differences in treatment-related adverse events or overall survival [114]. These results indicate that
transarterial chemoembolization and transarterial radioembolization are likely
safe and effective treatments for patients with MASH-HCC, yielding comparable
outcomes across different etiologies.
Systemic therapies: Phase III studies of systemic therapies in advanced HCC have
predominantly involved patients with compensated liver disease. However, the
etiology of liver disease has not been a consideration in treatment decisions or
trial designs. Typically, studies report efficacy based on stratification
factors such as etiology, often categorized as HBV, HCV, or
“non-viral.” The “non-viral” category includes
alcohol-related disease, MASH, and other causes (Table 1).
Table 1.
Summary of key phase III randomized trials evaluating the efficacy
and safety of systemic therapies according to the etiology of
hepatocellular carcinoma
Currently, several agents are approved for the first- and second-line treatment
of advanced HCC. These can be broadly categorized into two groups: multi-kinase
VEGFR-targeting small molecules and VEGFR2 monoclonal antibody approaches, as
well as immunotherapy-based approaches. Regarding overall survival, the efficacy
of the first group does not significantly vary based on the etiology of HCC, as
evidenced by similar HRs for overall survival in the study versus control arms.
This trend is also generally observed in secondary endpoints, such as
progression-free survival and objective response rates.
Unlike previous treatments, ICIs have not only demonstrated a survival benefit
but have also achieved significant response rates with durable responses lasting
over 20 months. There is growing interest in evaluating clinical characteristics
as markers of benefit, especially those associated with distinct pathogenic
pathways and immune profiles linked to different HCC etiologies. Two studies
have raised questions about the effectiveness of immunotherapies in
metabolic-associated steatohepatitis-HCC (MASH-HCC) compared to viral-related
HCC [72,115]. However, none of the phase III randomized controlled trials
(RCTs) in advanced HCC have reported the percentage of patients with MASH-HCC.
Consequently, indirect analysis of survival effects by etiology has been limited
to non-viral HCC cases. A meta-analysis of three RCTs (IMbrave150, CheckMate
459, and Keynote-240) indicated that patients with viral-related HCC responded
better to immunotherapies (HR, 0.64; 95% CI, 0.50–0.83) than those with
non-viral-related HCC (HR, 0.92; 95% CI, 0.77–1.11; P=0.2) [115]. Following the publication of a
subgroup analysis from the COSMIC-312 trial, a meta-analysis of four RCTs
confirmed a significant difference in efficacy (P=0.01) [116]. When the HIMALAYA trial, which assessed a
combination of two ICIs, was included in the meta-analysis (five RCTs), the
difference remained significant, albeit less pronounced (P=0.046) [15]. These findings suggest that
immunotherapies may be more effective in viral-related HCC than in other
etiologies, supporting observations that MASH-HCC tumors have dysfunctional T
cells, which may limit the effectiveness of ICIs [72].
However, these subgroup analyses are not statistically definitive and do not
account for other prognostic factors. The term "non-viral
etiologies" includes MASH-related, alcohol-related, idiopathic, and other
metabolic causes, which complicates the analysis. These findings suggest that
future studies should stratify participants based on etiology; however,
dedicated prospective studies are necessary to determine the specific role of
etiology. Although MASH-HCC is biologically distinct, the current clinical
approaches remain consistent with those used for other non-viral etiologies,
including alcohol-related HCC. Future trials should specifically identify cases
of MASH-related HCC to better understand the impact of immunotherapies on the
survival of this subgroup.
Conclusion
MASH is a significant global health issue and is projected to become the leading
cause of HCC by 2030. The progression from MASH to HCC is influenced by molecular
changes, the stage of fibrosis, the immune microenvironment, and the microbiome.
Lifestyle changes are crucial for preventing MASLD progression, and surveillance in
patients with MASH cirrhosis enables earlier detection and improves survival.
Currently, MASH-HCC is managed similarly to other HCC etiologies, but comorbidities
such as obesity and diabetes can complicate treatment.
Key unmet needs include identifying the molecular drivers of HCC in non-cirrhotic
MASLD and developing preventive therapies. There is also a need for improved
surveillance methods, particularly alternatives to ultrasound for obese patients,
and for refining the selection of surgical candidates. It is crucial to report
MASH-HCC outcomes separately in trials to facilitate better analysis; thus, it is
recommended that MASH-HCC be specifically identified in clinical trials to enable
more effective, personalized treatments. Additionally, further studies are required
to understand MASH-HCC-related T-cell dysfunction and to identify biomarkers that
predict treatment responses [117].
Authors' contributions
All work was done by Han Ah Lee.
Conflict of interest
No potential conflict of interest relevant to this article was reported.
Funding
This research was supported in part by a National Research Foundation of Korea
(NRF) grant funded by the Ministry of Science and ICT (grant no.
2022R1I1A1A01065244).
Data availability
Not applicable.
Acknowledgments
Not applicable.
Supplementary materials
Not applicable.
References
1. Kim DY. Changing etiology and epidemiology of hepatocellular carcinoma:
Asia and worldwide. J Liver Cancer 2024;24(1):62-70.
2. Korean Liver Cancer Association [KLCA], National Cancer Center [NCC]
Korea. 2022 KLCA-NCC Korea practice guidelines for the management of
hepatocellular carcinoma. J Liver Cancer 2023;23(1):1-120.
3. Lee HH, Lee HA, Kim EJ, Kim HY, Kim HC, Ahn SH, et al. Metabolic dysfunction-associated steatotic liver disease and risk
of cardiovascular disease. Gut 2024;73(3):533-540.
5. Anstee QM, Reeves HL, Kotsiliti E, Govaere O, Heikenwalder M. From NASH to HCC: current concepts and future
challenges. Nat Rev Gastroenterol Hepatol 2019;16(7):411-428.
7. Kim GA, Moon JH, Kim W. Critical appraisal of metabolic dysfunction-associated steatotic
liver disease: implication of Janus-faced modernity. Clin Mol Hepatol 2023;29(4):831-843.
8. Le MH, Le DM, Baez TC, Dang H, Nguyen VH, Lee K, et al. Global incidence of adverse clinical events in non-alcoholic
fatty liver disease: a systematic review and meta-analysis. Clin Mol Hepatol 2024;30(2):235-246.
9. Han JW, Sohn W, Choi GH, Jang JW, Seo GH, Kim BH, et al. Evolving trends in treatment patterns for hepatocellular
carcinoma in Korea from 2008 to 2022: a nationwide population-based
study. J Liver Cancer 2024;24(2):274-285.
11. Stine JG, Wentworth BJ, Zimmet A, Rinella ME, Loomba R, Caldwell SH, et al. Systematic review with meta-analysis: risk of hepatocellular
carcinoma in non-alcoholic steatohepatitis without cirrhosis compared to
other liver diseases. Aliment Pharmacol Ther 2018;48(7):696-703.
12. Kim DH. Combination of interventional oncology local therapies and
immunotherapy for the treatment of hepatocellular carcinoma. J Liver Cancer 2022;22(2):93-102.
14. Llovet JM, Heikenwalder M. Atezolizumab plus bevacizumab in advanced HCC: efficacy in
NASH-specific etiology. Gastroenterology 2023;165(5):1308-1310.
16. Dyson J, Jaques B, Chattopadyhay D, Lochan R, Graham J, Das D, et al. Hepatocellular cancer: the impact of obesity, type 2 diabetes and
a multidisciplinary team. J Hepatol 2014;60(1):110-117.
17. Estes C, Anstee QM, Arias-Loste MT, Bantel H, Bellentani S, Caballeria J, et al. Modeling NAFLD disease burden in China, France, Germany, Italy,
Japan, Spain, United Kingdom, and United States for the period
2016–2030. J Hepatol 2018;69(4):896-904.
18. White DL, Kanwal F, El–Serag HB. Association between nonalcoholic fatty liver disease and risk for
hepatocellular cancer, based on systematic review. Clin Gastroenterol Hepatol 2012;10(12):1342-1359.E2.
19. Yang JD, Ahmed F, Mara KC, Addissie BD, Allen AM, Gores GJ, et al. Diabetes is associated with increased risk of hepatocellular
carcinoma in patients with cirrhosis from nonalcoholic fatty liver
disease. Hepatology 2020;71(3):907-916.
20. Kanwal F, Kramer JR, Mapakshi S, Natarajan Y, Chayanupatkul M, Richardson PA, et al. Risk of hepatocellular cancer in patients with non-alcoholic
fatty liver disease. Gastroenterology 2018;155(6):1828-1837.E2.
21. Mittal S, El-Serag HB, Sada YH, Kanwal F, Duan Z, Temple S, et al. Hepatocellular carcinoma in the absence of cirrhosis in United
States Veterans is associated with nonalcoholic fatty liver
disease. Clin Gastroenterol Hepatol 2016;14(1):124-131.E1.
22. Alexander M, Loomis AK, van der Lei J, Duarte-Salles T, Prieto-Alhambra D, Ansell D, et al. Risks and clinical predictors of cirrhosis and hepatocellular
carcinoma diagnoses in adults with diagnosed NAFLD: real-world study of 18
million patients in four European cohorts. BMC Med 2019;17(1):95
23. Kanwal F, Kramer JR, Li L, Dai J, Natarajan Y, Yu X, et al. Effect of metabolic traits on the risk of cirrhosis and
hepatocellular cancer in nonalcoholic fatty liver disease. Hepatology 2020;71(3):808-819.
24. Rustgi VK, Li Y, Gupta K, Minacapelli CD, Bhurwal A, Catalano C, et al. Bariatric surgery reduces cancer risk in adults with nonalcoholic
fatty liver disease and severe obesity. Gastroenterology 2021;161(1):171-184.E10.
25. Chang Y, Cho YK, Kim Y, Sung E, Ahn J, Jung HS, et al. Nonheavy drinking and worsening of noninvasive fibrosis markers
in nonalcoholic fatty liver disease: a cohort study. Hepatology 2019;69(1):64-75.
26. Ascha MS, Hanouneh IA, Lopez R, Tamimi TAR, Feldstein AF, Zein NN. The incidence and risk factors of hepatocellular carcinoma in
patients with nonalcoholic steatohepatitis. Hepatology 2010;51(6):1972-1978.
27. Kimura T, Tanaka N, Fujimori N, Sugiura A, Yamazaki T, Joshita S, et al. Mild drinking habit is a risk factor for hepatocarcinogenesis in
non-alcoholic fatty liver disease with advanced fibrosis. World J Gastroenterol 2018;24(13):1440-1450.
28. Abdel-Rahman O, Helbling D, Schöb O, Eltobgy M, Mohamed H, Schmidt J, et al. Cigarette smoking as a risk factor for the development of and
mortality from hepatocellular carcinoma: an updated systematic review of 81
epidemiological studies. J Evid Based Med 2017;10(4):245-254.
29. Cavin C, Holzhaeuser D, Scharf G, Constable A, Huber WW, Schilter B. Cafestol and kahweol, two coffee specific diterpenes with
anticarcinogenic activity. Food Chem Toxicol 2002;40(8):1155-1163.
30. Majer BJ, Hofer E, Cavin C, Lhoste E, Uhl M, Glatt HR, et al. Coffee diterpenes prevent the genotoxic effects of
2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP) and
N-nitrosodimethylamine in a human derived liver cell line
(HepG2). Food Chem Toxicol 2005;43(3):433-441.
31. Huxley R, Lee CMY, Barzi F, Timmermeister L, Czernichow S, Perkovic V, et al. Coffee, decaffeinated coffee, and tea consumption in relation to
incident type 2 diabetes mellitus: a systematic review with
meta-analysis. Arch Intern Med 2009;169(22):2053-2063.
32. Zheng L, Yang W, Wu F, Wang C, Yu L, Tang L, et al. Prognostic significance of AMPK activation and therapeutic
effects of metformin in hepatocellular carcinoma. Clin Cancer Res 2013;19(19):5372-5380.
33. Buzzai M, Jones RG, Amaravadi RK, Lum JJ, DeBerardinis RJ, Zhao F, et al. Systemic treatment with the antidiabetic drug metformin
selectively impairs p53-deficient tumor cell growth. Cancer Res 2007;67(14):6745-6752.
34. Forslund K, Hildebrand F, Nielsen T, Falony G, Le Chatelier E, Sunagawa S, et al. Disentangling type 2 diabetes and metformin treatment signatures
in the human gut microbiota. Nature 2015;528(7581):262-266.
35. Cao Z, Fan-Minogue H, Bellovin DI, Yevtodiyenko A, Arzeno J, Yang Q, et al. MYC phosphorylation, activation, and tumorigenic potential in
hepatocellular carcinoma are regulated by HMG-CoA reductase. Cancer Res 2011;71(6):2286-2297.
36. Kramer JR, Natarajan Y, Dai J, Yu X, Li L, El‐Serag HB, et al. Effect of diabetes medications and glycemic control on risk of
hepatocellular cancer in patients with nonalcoholic fatty liver
disease. Hepatology 2022;75(6):1420-1428.
37. Lee TY, Wu JC, Yu SH, Lin JT, Wu MS, Wu CY. The occurrence of hepatocellular carcinoma in different risk
stratifications of clinically noncirrhotic nonalcoholic fatty liver
disease. Int J Cancer 2017;141(7):1307-1314.
38. Mao X, Zhang X, Kam L, Chien N, Lai R, Cheung KS, et al. Synergistic association of sodium-glucose cotransporter-2
inhibitor and metformin on liver and non-liver complications in patients
with type 2 diabetes mellitus and metabolic dysfunction-associated steatotic
liver disease. Gut 2024 Aug 8 [Epub].
39. Roudier E, Mistafa O, Stenius U. Statins induce mammalian target of rapamycin (mTOR)-mediated
inhibition of Akt signaling and sensitize p53-deficient cells to cytostatic
drugs. Mol Cancer Ther 2006;5(11):2706-2715.
40. Relja B, Meder F, Wang M, Blaheta R, Henrich D, Marzi I, et al. Simvastatin modulates the adhesion and growth of hepatocellular
carcinoma cells via decrease of integrin expression and ROCK. Int J Oncol 2011;38(3):879-885.
41. Sutter AP, Maaser K, Höpfner M, Huether A, Schuppan D, Scherübl H. Cell cycle arrest and apoptosis induction in hepatocellular
carcinoma cells by HMG-CoA reductase inhibitors. Synergistic
antiproliferative action with ligands of the peripheral benzodiazepine
receptor. J Hepatol 2005;43(5):808-816.
42. Kah J, Wüstenberg A, Keller AD, Sirma H, Montalbano R, Ocker M, et al. Selective induction of apoptosis by HMG-CoA reductase inhibitors
in hepatoma cells and dependence on p53 expression. Oncol Rep 2012;28(3):1077-1083.
43. German MN, Lutz MK, Pickhardt PJ, Bruce RJ, Said A. Statin use is protective against hepatocellular carcinoma in
patients with nonalcoholic fatty liver disease: a case-control
study. J Clin Gastroenterol 2020;54(8):733-740.
44. Pinyopornpanish K, Al-Yaman W, Butler RS, Carey W, McCullough A, Romero-Marrero C. Chemopreventive effect of statin on hepatocellular carcinoma in
patients with nonalcoholic steatohepatitis cirrhosis. Am J Gastroenterol 2021;116(11):2258-2269.
45. Vilar-Gomez E, Calzadilla-Bertot L, Wai-Sun Wong V, Castellanos M, Aller-de la Fuente R, Metwally M, et al. Fibrosis severity as a determinant of cause-specific mortality in
patients with advanced nonalcoholic fatty liver disease: a multi-national
cohort study. Gastroenterology 2018;155(2):443-457.E17.
47. Fuchs A, Samovski D, Smith GI, Cifarelli V, Farabi SS, Yoshino J, et al. Associations among adipose tissue immunology, inflammation,
exosomes and insulin sensitivity in people with obesity and nonalcoholic
fatty liver disease. Gastroenterology 2021;161(3):968-981.E12.
49. Daugherity EK, Balmus G, Al Saei A, Moore ES, Abi Abdallah D, Rogers AB, et al. The DNA damage checkpoint protein ATM promotes hepatocellular
apoptosis and fibrosis in a mouse model of non-alcoholic fatty liver
disease. Cell Cycle 2012;11(10):1918-1928.
50. Ng SWK, Rouhani FJ, Brunner SF, Brzozowska N, Aitken SJ, Yang M, et al. Convergent somatic mutations in metabolism genes in chronic liver
disease. Nature 2021;598(7881):473-478.
51. Zhu C, Kim K, Wang X, Bartolome A, Salomao M, Dongiovanni P, et al. Hepatocyte notch activation induces liver fibrosis in
nonalcoholic steatohepatitis. Sci Transl Med 2018;10(468):eaat0344
52. Zhu C, Tabas I, Schwabe RF, Pajvani UB. Maladaptive regeneration: the reawakening of developmental
pathways in NASH and fibrosis. Nat Rev Gastroenterol Hepatol 2021;18(2):131-142.
54. Sanyal AJ, Van Natta ML, Clark J, Neuschwander-Tetri BA, Diehl A, Dasarathy S, et al. Prospective study of outcomes in adults with nonalcoholic fatty
liver disease. N Engl J Med 2021;385(17):1559-1569.
55. Ramachandran P, Dobie R, Wilson-Kanamori JR, Dora EF, Henderson BEP, Luu NT, et al. Resolving the fibrotic niche of human liver cirrhosis at
single-cell level. Nature 2019;575(7783):512-518.
57. Lei H, Reinke P, Volk HD, Lv Y, Wu R. Mechanisms of immune tolerance in liver transplantation-crosstalk
between alloreactive T cells and liver cells with therapeutic
prospects. Front Immunol 2019;10:2667
58. Kitade M, Yoshiji H, Kojima H, Ikenaka Y, Noguchi R, Kaji K, et al. Neovascularization and oxidative stress in the progression of
non-alcoholic steatohepatitis. Mol Med Rep 2008;1(4):543-548.
59. Coulon S, Legry V, Heindryckx F, Van Steenkiste C, Casteleyn C, Olievier K, et al. Role of vascular endothelial growth factor in the pathophysiology
of nonalcoholic steatohepatitis in two rodent models. Hepatology 2013;57(5):1793-1805.
60. Kwak M, Mehaffey JH, Hawkins RB, Hsu A, Schirmer B, Hallowell PT. Bariatric surgery is associated with reduction in non-alcoholic
steatohepatitis and hepatocellular carcinoma: a propensity matched
analysis. Am J Surg 2020;219(3):504-507.
61. Yan M, Man S, Ma L, Guo L, Huang L, Gao W. Immunological mechanisms in steatotic liver diseases: an overview
and clinical perspectives. Clin Mol Hepatol 2024 Jul 11 [Epub].
63. Wolf MJ, Adili A, Piotrowitz K, Abdullah Z, Boege Y, Stemmer K, et al. Metabolic activation of intrahepatic CD8+ T cells and
NKT cells causes nonalcoholic steatohepatitis and liver cancer via
cross-talk with hepatocytes. Cancer Cell 2014;26(4):549-564.
64. Heinrich B, Brown ZJ, Diggs LP, Vormehr M, Ma C, Subramanyam V, et al. Steatohepatitis impairs T-cell–directed immunotherapies
against liver tumors in mice. Gastroenterology 2021;160(1):331-345.E6.
65. Ma C, Kesarwala AH, Eggert T, Medina-Echeverz J, Kleiner DE, Jin P, et al. NAFLD causes selective CD4+ T lymphocyte loss and
promotes hepatocarcinogenesis. Nature 2016;531(7593):253-257.
66. Deczkowska A, David E, Ramadori P, Pfister D, Safran M, Li B, et al. XCR1+ type 1 conventional dendritic cells drive liver
pathology in non-alcoholic steatohepatitis. Nat Med 2021;27(6):1043-1054.
67. Ou R, Liu J, Lv M, Wang J, Wang J, Zhu L, et al. Neutrophil depletion improves diet-induced non-alcoholic fatty
liver disease in mice. Endocrine 2017;57(1):72-82.
69. McVey JC, Green BL, Ruf B, McCallen JD, Wabitsch S, Subramanyam V, et al. NAFLD indirectly impairs antigen-specific CD8+ T cell
immunity against liver cancer in mice. iScience 2022;25(2):103847
71. Dudek M, Pfister D, Donakonda S, Filpe P, Schneider A, Laschinger M, et al. Auto-aggressive CXCR6+ CD8 T cells cause liver immune
pathology in NASH. Nature 2021;592(7854):444-449.
72. Pfister D, Núñez NG, Pinyol R, Govaere O, Pinter M, Szydlowska M, et al. NASH limits anti-tumour surveillance in immunotherapy-treated
HCC. Nature 2021;592(7854):450-456.
73. Ma C, Han M, Heinrich B, Fu Q, Zhang Q, Sandhu M, et al. Gut microbiome–mediated bile acid metabolism regulates
liver cancer via NKT cells. Science 2018;360(6391):eaan5931
74. Jadhav K, Cohen TS. Can you trust your gut? Implicating a disrupted intestinal
microbiome in the progression of NAFLD/NASH. Front Endocrinol 2020;11:592157
76. Mouries J, Brescia P, Silvestri A, Spadoni I, Sorribas M, Wiest R, et al. Microbiota-driven gut vascular barrier disruption is a
prerequisite for non-alcoholic steatohepatitis development. J Hepatol 2019;71(6):1216-1228.
77. Boursier J, Mueller O, Barret M, Machado M, Fizanne L, Araujo‐Perez F, et al. The severity of nonalcoholic fatty liver disease is associated
with gut dysbiosis and shift in the metabolic function of the gut
microbiota. Hepatology 2016;63(3):764-775.
78. Madrid AM, Hurtado C, Venegas M, Cumsille F, Defilippi C. Long-term treatment with cisapride and antibiotics in liver
cirrhosis: effect on small intestinal motility, bacterial overgrowth, and
liver function. Am J Gastroenterol 2001;96(4):1251-1255.
82. Kawaguchi T, Shima T, Mizuno M, Mitsumoto Y, Umemura A, Kanbara Y, et al. Risk estimation model for nonalcoholic fatty liver disease in the
Japanese using multiple genetic markers. PLoS One 2018;13(1):e0185490.
84. Hoshida Y, Nijman SMB, Kobayashi M, Chan JA, Brunet JP, Chiang DY, et al. Integrative transcriptome analysis reveals common molecular
subclasses of human hepatocellular carcinoma. Cancer Res 2009;69(18):7385-7392.
85. Pinyol R, Torrecilla S, Wang H, Montironi C, Piqué-Gili M, Torres-Martin M, et al. Molecular characterisation of hepatocellular carcinoma in
patients with non-alcoholic steatohepatitis. J Hepatol 2021;75(4):865-878.
86. Jühling F, Hamdane N, Crouchet E, Li S, El Saghire H, Mukherji A, et al. Targeting clinical epigenetic reprogramming for chemoprevention
of metabolic and viral hepatocellular carcinoma. Gut 2021;70(1):157-169.
87. Singh S, Singh PP, Singh AG, Murad MH, Sanchez W. Statins are associated with a reduced risk of hepatocellular
cancer: a systematic review and meta-analysis. Gastroenterology 2013;144(2):323-332.
88. Simon TG, Duberg AS, Aleman S, Chung RT, Chan AT, Ludvigsson JF. Association of aspirin with hepatocellular carcinoma and
liver-related mortality. N Engl J Med 2020;382(11):1018-1028.
89. European Association for the Study of the Liver [EASL], European
Association for the Study of Diabetes [EASD], European Association for
the Study of Obesity [EASO]. EASL–EASD–EASO clinical practice guidelines on the
management of metabolic dysfunction-associated steatotic liver disease
(MASLD). J Hepatol 2024;81(3):492-542.
90. Kang SH, Lee HW, Yoo JJ, Cho Y, Kim SU, Lee TH, et al. KASL clinical practice guidelines: management of nonalcoholic
fatty liver disease. Clin Mol Hepatol 2021;27(3):363-401.
92. Baumeister SE, Schlesinger S, Aleksandrova K, Jochem C, Jenab M, Gunter MJ, et al. Association between physical activity and risk of hepatobiliary
cancers: a multinational cohort study. J Hepatol 2019;70(5):885-892.
93. Promrat K, Kleiner DE, Niemeier HM, Jackvony E, Kearns M, Wands JR, et al. Randomized controlled trial testing the effects of weight loss on
nonalcoholic steatohepatitis. Hepatology 2010;51(1):121-129.
95. Aby E, Phan J, Truong E, Grotts J, Saab S. Inadequate hepatocellular carcinoma screening in patients with
nonalcoholic steatohepatitis cirrhosis. J Clin Gastroenterol 2019;53(2):142-146.
96. Wolf E, Rich NE, Marrero JA, Parikh ND, Singal AG. Use of hepatocellular carcinoma surveillance in patients with
cirrhosis: a systematic review and meta‐analysis. Hepatology 2021;73(2):713-725.
97. Tan DJH, Ng CH, Lin SY, Pan XH, Tay P, Lim WH, et al. Clinical characteristics, surveillance, treatment allocation, and
outcomes of non-alcoholic fatty liver disease-related hepatocellular
carcinoma: a systematic review and meta-analysis. Lancet Oncol 2022;23(4):521-530.
98. Chong N, Schoenberger H, Yekkaluri S, Fetzer DT, Rich NE, Yokoo T, et al. Association between ultrasound quality and test performance for
HCC surveillance in patients with cirrhosis: a retrospective cohort
study. Aliment Pharmacol Ther 2022;55(6):683-690.
100. Tzartzeva K, Obi J, Rich NE, Parikh ND, Marrero JA, Yopp A, et al. Surveillance imaging and alpha fetoprotein for early detection of
hepatocellular carcinoma in patients with cirrhosis: a
meta-analysis. Gastroenterology 2018;154(6):1706-1718.e1.
101. Yu JH, Lee HA, Kim SU. Noninvasive imaging biomarkers for liver fibrosis in nonalcoholic
fatty liver disease: current and future. Clin Mol Hepatol 2023;29(Suppl):S136-S149.
102. Koh X, Tan J, Liew X, Syn N, Teo Y, Lee Y, et al. Liver resection for nonalcoholic fatty liver disease-associated
hepatocellular carcinoma. J Am Coll Surg 2019;229(5):467-478e1.
103. Foerster F, Gairing SJ, Müller L, Galle PR. NAFLD-driven HCC: safety and efficacy of current and emerging
treatment options. J Hepatol 2022;76(2):446-457.
104. Petrelli F, Cortellini A, Indini A, Tomasello G, Ghidini M, Nigro O, et al. Association of obesity with survival outcomes in patients with
cancer: a systematic review and meta-analysis. JAMA Netw Open 2021;4(3):e213520.
105. Wang YG, Wang P, Wang B, Fu ZJ, Zhao WJ, Yan SL. Diabetes mellitus and poorer prognosis in hepatocellular
carcinoma: a systematic review and meta-analysis. PLoS One 2014;9(5):e95485.
106. Su CW, Chau GY, Hung HH, Yeh YC, Lei HJ, Hsia CY, et al. Impact of steatosis on prognosis of patients with early-stage
hepatocellular carcinoma after hepatic resection. Ann Surg Oncol 2015;22(7):2253-2261.
107. Molinari M, Kaltenmeier C, Samra PB, Liu H, Wessel C, Lou Klem M, et al. Hepatic resection for hepatocellular carcinoma in nonalcoholic
fatty liver disease: a systematic review and meta-analysis of 7226
patients. Ann Surg Open 2021;2(2):e065.
108. Chin KM, Prieto M, Cheong CK, Di Martino M, Ielpo B, Goh BKP, et al. Outcomes after curative therapy for hepatocellular carcinoma in
patients with non-alcoholic fatty liver disease: a meta-analysis and review
of current literature. HPB 2021;23(8):1164-1174.
109. Wong RJ, Chou C, Bonham CA, Concepcion W, Esquivel CO, Ahmed A. Improved survival outcomes in patients with non-alcoholic
steatohepatitis and alcoholic liver disease following liver transplantation:
an analysis of 2002–2012 United Network for Organ Sharing
data. Clin Transplant 2014;28(6):713-721.
111. Kern B, Feurstein B, Fritz J, Fabritius C, Sucher R, Graziadei I, et al. High incidence of hepatocellular carcinoma and postoperative
complications in patients with nonalcoholic steatohepatitis as a primary
indication for deceased liver transplantation. Eur J Gastroenterol Hepatol 2019;31(2):205-210.
112. Wong CR, Njei B, Nguyen MH, Nguyen A, Lim JK. Survival after treatment with curative intent for hepatocellular
carcinoma among patients with vs without non-alcoholic fatty liver
disease. Aliment Pharmacol Ther 2017;46(11-12):1061-1069.
113. Young S, Sanghvi T, Rubin N, Hall D, Roller L, Charaf Y, et al. Transarterial chemoembolization of hepatocellular carcinoma:
propensity score matching study comparing survival and complications in
patients with nonalcoholic steatohepatitis versus other causes
cirrhosis. Cardiovasc Intervent Radiol 2020;43(1):65-75.
114. Schotten C, Bechmann LP, Manka P, Theysohn J, Dechêne A, El Fouly A, et al. NAFLD-associated comorbidities in advanced stage HCC do not alter
the safety and efficacy of yttrium-90 radioembolization. Liver Cancer 2019;8(6):491-504.
118. Finn RS, Qin S, Ikeda M, Galle PR, Ducreux M, Kim TY, et al. Atezolizumab plus bevacizumab in unresectable hepatocellular
carcinoma. N Engl J Med 2020;382(20):1894-1905.
119. Yau T, Kaseb A, Cheng AL, Qin S, Zhu AX, Chan SL, et al. Cabozantinib plus atezolizumab versus sorafenib for advanced
hepatocellular carcinoma (COSMIC-312): final results of a randomised phase 3
study. Lancet Gastroenterol Hepatol 2024;9(4):310-322.
120. Abou-Alfa GK, Lau G, Kudo M, Chan SL, Kelley RK, Furuse J, et al. Tremelimumab plus durvalumab in unresectable hepatocellular
carcinoma. NEJM Evid 2022;1(8):EVIDoa2100070
122. Qin S, Kudo M, Meyer T, Bai Y, Guo Y, Meng Z, et al. Tislelizumab vs sorafenib as first-line treatment for
unresectable hepatocellular carcinoma: a phase 3 randomized clinical
trial. JAMA Oncol 2023;9(12):1651-1659.
123. Llovet JM, Kudo M, Merle P, Meyer T, Qin S, Ikeda M, et al. Lenvatinib plus pembrolizumab versus lenvatinib plus placebo for
advanced hepatocellular carcinoma (LEAP-002): a randomised, double-blind,
phase 3 trial. Lancet Oncol 2023;24(12):1399-1410.
124. Qin S, Chan SL, Gu S, Bai Y, Ren Z, Lin X, et al. Camrelizumab plus rivoceranib versus sorafenib as first-line
therapy for unresectable hepatocellular carcinoma (CARES-310): a randomised,
open-label, international phase 3 study. Lancet 2023;402(10408):1133-1146.
125. Ren Z, Xu J, Bai Y, Xu A, Cang S, Du C, et al. Sintilimab plus a bevacizumab biosimilar (IBI305) versus
sorafenib in unresectable hepatocellular carcinoma (ORIENT-32): a
randomised, open-label, phase 2–3 study. Lancet Oncol 2021;22(7):977-990.
126. Finn RS, Ryoo BY, Merle P, Kudo M, Bouattour M, Lim HY, et al. Pembrolizumab as second-line therapy in patients with advanced
hepatocellular carcinoma in KEYNOTE-240: a randomized, double-blind, phase
III trial. J Clin Oncol 2020;38(3):193-202.
127. Qin S, Chen Z, Fang W, Ren Z, Xu R, Ryoo BY, et al. Pembrolizumab versus placebo as second-line therapy in patients
from Asia with advanced hepatocellular carcinoma: a randomized,
double-blind, phase III trial. J Clin Oncol 2023;41(7):1434-1443.
128. Castet F, Willoughby CE, Haber PK, Llovet JM. Atezolizumab plus bevacizumab: a novel breakthrough in
hepatocellular carcinoma. Clin Cancer Res 2021;27(7):1827-1829.
129. Bruix J, Raoul JL, Sherman M, Mazzaferro V, Bolondi L, Craxi A, et al. Efficacy and safety of sorafenib in patients with advanced
hepatocellular carcinoma: subanalyses of a phase III trial. J Hepatol 2012;57(4):821-829.
130. Cheng AL, Kang YK, Chen Z, Tsao CJ, Qin S, Kim JS, et al. Efficacy and safety of sorafenib in patients in the Asia-Pacific
region with advanced hepatocellular carcinoma: a phase III randomised,
double-blind, placebo-controlled trial. Lancet Oncol 2009;10(1):25-34.
131. Cheng AL, Guan Z, Chen Z, Tsao CJ, Qin S, Kim JS, et al. Efficacy and safety of sorafenib in patients with advanced
hepatocellular carcinoma according to baseline status: subset analyses of
the phase III sorafenib Asia–Pacific trial. Eur J Cancer 2012;48(10):1452-1465.
132. Kudo M, Finn RS, Qin S, Han KH, Ikeda K, Piscaglia F, et al. Lenvatinib versus sorafenib in first-line treatment of patients
with unresectable hepatocellular carcinoma: a randomised phase 3
non-inferiority trial. Lancet 2018;391(10126):1163-1173.
133. Abou-Alfa GK, Meyer T, Cheng AL, El-Khoueiry AB, Rimassa L, Ryoo BY, et al. Cabozantinib in patients with advanced and progressing
hepatocellular carcinoma. N Engl J Med 2018;379(1):54-63.
135. Zhu AX, Kang YK, Yen CJ, Finn RS, Galle PR, Llovet JM, et al. Ramucirumab after sorafenib in patients with advanced
hepatocellular carcinoma and increased α-fetoprotein concentrations
(REACH-2): a randomised, double-blind, placebo-controlled, phase 3
trial. Lancet Oncol 2019;20(2):282-296.
From HBV to MASLD Cirrhosis: Mechanistic Insights and Therapeutic Strategies Hanqi Yu, Hongfan Ding, Yang Huang, Haoze Cao, Yuan Ding, Weilin Wang Portal Hypertension & Cirrhosis.2026; 5(2): 170. CrossRef
Assessing hepatocellular carcinoma in Nigeria: A multicenter retrospective cohort review of prevalence, risk factors, and treatment outcomes Yusuf Musa, Nasiru Altine Dankiri, Habib Tijjani Saleh, Chinwe Philomena Onyia, Lukman Olaitan Abdulkareem, Kenechukwu C Okonkwo, Fatimah Biade Abdulkareem, Rukayya Babale Shu'aibu, Abubakar Sadiq Aminu, Adamu Mohammad Ewa, Hafizu Zubairu Abdullahi, Adiri World Journal of Gastrointestinal Oncology.2026;[Epub] CrossRef
Network Pharmacology and In Vivo Validation Reveal Berberine-Mediated Regulation of the Liver–Brain Inflammatory Axis in MCD-Induced Steatohepatitis Yeon-Joo Yoo, Ji-Han Kim, Seung-Hoon Yoo, Byung-Cheol Lee International Journal of Molecular Sciences.2026; 27(15): 6967. CrossRef
The MASLD–Cardio-Oncology Triangle: Dietary Patterns, Metabolic Remodelling and Implications for Cancer Therapy Tolerance Francesca La Rocca, Graziella Privitera, Calogero Geraci, Valentina Morello, Giulio Geraci, Valentina Paternò, Ciro Santoro, Roberta Esposito Nutrients.2026; 18(15): 2527. CrossRef
Pathogenesis and management of metabolic dysfunction-associated
steatohepatitis-related hepatocellular carcinoma: a narrative
review
Fig. 1.
Pathogenesis and progression of MASH-HCC (drawn by the author). MASH,
metabolic dysfunction-associated steatohepatitis; HCC, hepatocellular
carcinoma; TH, T helper.
Fig. 1.
Pathogenesis and management of metabolic dysfunction-associated
steatohepatitis-related hepatocellular carcinoma: a narrative
review
Summary of key phase III randomized trials evaluating the efficacy
and safety of systemic therapies according to the etiology of
hepatocellular carcinoma
Table 1.
Summary of key phase III randomized trials evaluating the efficacy
and safety of systemic therapies according to the etiology of
hepatocellular carcinoma